Recently in 2025, we developed a new local correlation formalism
1431
J. Chem. Theory Comput.
(2023),
19,
pp. 7577.
Link
,
1434
J. Chem. Theory Comput.
(2025),
21,
pp. 6887.
Link
,
1433
J. Chem. Theory Comput.
(2025),
21,
pp. 10910.
Link
based on numerical sparsity. The goal for this new formalism is to be as
close as the canonical RI-MP2 as possible, with the consideration of only
having one user input parameter for desired accuracy.
Right now, the code works for elements up to xenon for restricted and unrestricted SCF references.
The MP2 energy is
| (6.19) |
where (and the exchange-like tensor ) is the electron repulsion integral (ERI) tensor
| (6.20) |
is the solution to the MP2 equation
| (6.21) |
which is a set of linear equations and with orthogonal orbitals, is
| (6.22) |
We compute and store the tensor in a numerical sparsity way, meaning that for each significant pair, we store two "ragged lists" of significant indeces and their integral values. The main approximation made by us is the "fixed sparsity pattern approximation", which means we fixed the solution to have the same sparsity pattern (same list of significant indices), see Fig 6.1.
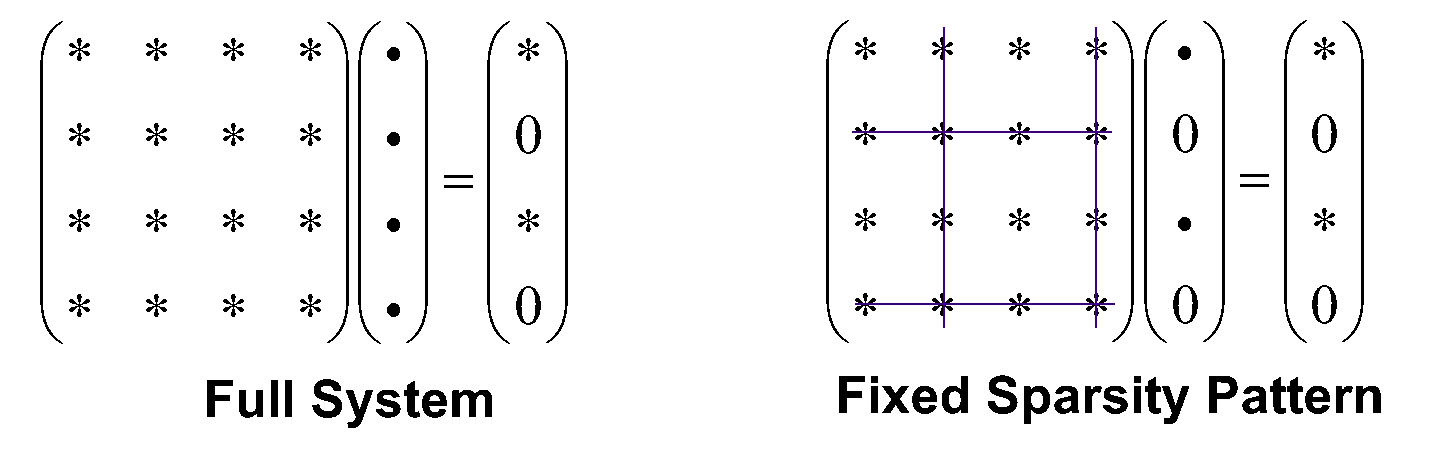
The code is called by setting LMP2 option for CORRELATION. Localization must be specified in LOCOOCCVIRT. AO linear dependency is treated differently for different localization schemes.
LOCOOCCVIRT
LOCOOCCVIRT
Localizes MO orbitals.
TYPE:
INTEGER
DEFAULT:
1
Normal AO linear dependency treatment. This will NOT work with VV-HV when AO are linearly dependent.
OPTIONS:
2
Repeatedly remove basis functions when linear dependency is present. This will CHANGE SCF result but work with VV-HV.
RECOMMENDATION:
Use 2 since VV-HV is preferred. The reason that a non-default is recommended is that
it will result in different SCF energies.
Different methods for localizing orbitals can be chosen. -Boys is recommended for the occupied space. -Boys tries to minimize the total orbital spread and the total off-diagonal Fock at the same time.
| (6.23) |
Boys orbitals (minimizing the total orbital spread) are obtained if , whereas canonical orbitals (minimizing off-diagonal elements of the Fock matrix) are obtained as . The value is recommended.
method
Defines the method to localize the occupied space.
INPUT SECTION: $localocc
TYPE:
STRING
DEFAULT:
newboys
OPTIONS:
newboys, cholesky, pipekmezey
RECOMMENDATION:
Use the default since -Boys orbitals are
shown to be very effective.
lambda
Defines .
INPUT SECTION: $localocc
TYPE:
FLOAT
DEFAULT:
0
OPTIONS:
100
Preferred parameter
RECOMMENDATION:
Using larger will decrease the accuracy of
the local MP2 code.
VV-HV is the recommended localization method for the virtual space.
method
Defines the method to localize the virtual space.
INPUT SECTION: $localvirt
TYPE:
STRING
DEFAULT:
VV-HV
VV-HV orbitals
OPTIONS:
VV-HV, NewBoys, PipekMezey, PAO
RECOMMENDATION:
Use the default.
Thresholds control the accuracy-efficiency tradeoff. Normally, the user only need to specify the main threshold (), to control what elements to retain in .
thresh
Defines the main threshold .
INPUT SECTION: $localmp2
TYPE:
INTEGER
DEFAULT:
60
OPTIONS:
55 (, close to VeryTight in DLPNO-MP2), 50, 60, 65, 70
RECOMMENDATION:
The default typically recovers 99.997% correlation energy
and has 0.1 kcalmol energy difference error.
The error scales linearly with , meaning that using 50
will accelerate the code by 3 to 5 times but the error will also be 10 bigger.
All other thresholds for inner prescreening are listed here, they can be changed similarly. The default pre-screener threshold is
| (6.24) |
| Threshold | Handle | Default | Note |
| thresh | user specified main parameter | ||
| threshuFERF | |||
| threshia | Eq. 6.24 | ||
| threshiP | |||
| threshcoeffocc | |||
| threshcoeffvirt | fixed default | ||
| threshsol | residue in the linear equation solver | ||
| threshpreia | pre-screeners for computing pre-screener | ||
| threshpreiP | |||
| threshpremuP |
-MP2 and second-order size-consistent Brillouin-Wigner theory (BWs2, Section 6.8) are also supported but cannot both be enabled at the same time.
regularize
Defines in -MP2.
INPUT SECTION: $localmp2
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
1450
RECOMMENDATION:
is recommended for non-covalent interactions.
bw
Enables BWs2 theory.
INPUT SECTION: $localmp2
TYPE:
INTEGER
DEFAULT:
0
disabled
OPTIONS:
1
enabled
RECOMMENDATION:
Use 1 if BW is needed.
bw_alpha
Defines parameter in BWs2.
INPUT SECTION: $localmp2
TYPE:
INTEGER
DEFAULT:
1000
is exact for two-electron in two-orbital problem.
OPTIONS:
4000
.
RECOMMENDATION:
is recommended for best chemical energy differences.
Together with , is recommended.
bw_gamma
Defines parameter in BWs2.
INPUT SECTION: $localmp2
TYPE:
INTEGER
DEFAULT:
0
.
OPTIONS:
2000
.
RECOMMENDATION:
is recommended for best chemistry.
Example 6.3 A closed-shell example with . For other cases, modify the geometry, basis sets, and threshold as needed.
$molecule 0 1 C1 H2 C1 1.09774 H3 C1 1.09776 H2 107.23372 H4 C1 1.09778 H2 107.22823 H3 114.89419 H5 C1 1.09778 H2 107.22823 H4 114.89419 $end $rem JOBTYPE¯¯¯SP UNRESTRICTED¯¯FALSE CORRELATION¯¯LMP2 LOCOOCCVIRT¯¯2 BASIS¯¯¯DEF2-SVP AUX_BASIS¯¯RIMP2-DEF2-SVP N_FROZEN_CORE¯FC SCF_CONVERGENCE 9 THRESH¯¯¯14 SYMMETRY¯¯FALSE SYM_IGNORE¯¯TRUE $end $localocc method newboys lambda 100 $end $localvirt method vv-hv $end $localmp2 thresh 60 $end