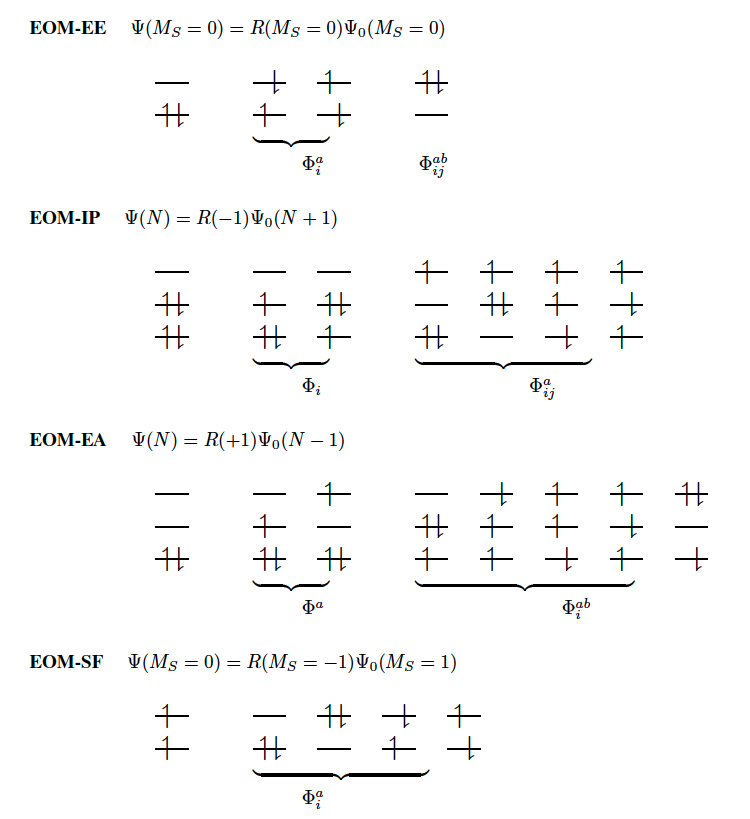
One can describe electronically excited states at a level of theory similar to that associated with coupled-cluster theory for the ground state by applying either linear response theory577 or equation-of-motion methods.1061 A number of groups have demonstrated that excitation energies based on a coupled-cluster singles and doubles ground state are generally very accurate for states that are primarily single electron promotions. The error observed in calculated excitation energies to such states is typically 0.1–0.2 eV, with 0.3 eV as a conservative estimate, including both valence and Rydberg excited states. This, of course, assumes that a basis set large and flexible enough to describe the valence and Rydberg states is employed. The accuracy of excited state coupled-cluster methods is much lower for excited states that involve a substantial double excitation character, where errors may be 1 eV or even more. Such errors arise because the description of electron correlation of an excited state with substantial double excitation character requires higher truncation of the excitation operator. The description of these states can be improved by including triple excitations, as in EOM(2,3).
Q-Chem includes coupled-cluster methods for excited states based on the coupled cluster singles and doubles (CCSD) method described earlier. CCMAN also includes the optimized orbital coupled-cluster doubles (OD) variant. OD excitation energies have been shown to be essentially identical in numerical performance to CCSD excited states.599
These methods, while far more computationally expensive than TDDFT, are nevertheless useful as proven high accuracy methods for the study of excited states of small molecules. Moreover, they are capable of describing both valence and Rydberg excited states, as well as states of a charge-transfer character. Also, when studying a series of related molecules it can be very useful to compare the performance of TDDFT and coupled-cluster theory for at least a small example to understand its performance. Along similar lines, the CIS(D) method described earlier as an economical correlation energy correction to CIS excitation energies is in fact an approximation to EOM-CCSD. It is useful to assess the performance of CIS(D) for a class of problems by benchmarking against the full coupled-cluster treatment. Finally, Q-Chem includes extensions of EOM methods to treat ionized or electron attachment systems, as well as di- and triradicals.