- 1 Introduction
- 2 Installation, Customization, and Execution
- 3 Q-Chem Inputs
- 3.1 IQmol
- 3.2 General Form
- 3.3 Molecular Coordinate Input ($molecule)
- 3.4 Cartesian Coordinates
- 3.5 Z-matrix Coordinates
- 3.6 Job Specification: The $rem Array Concept
- 3.7 $rem Array Format in Q-Chem Input
- 3.8 Minimum $rem Array Requirements
- 3.9 Comments ($comment)
- 3.10 User-Defined Basis Sets ($basis and $aux_basis)
- 3.11 User-Defined Pseudopotentials ($ecp)
- 3.12 User-Defined Exchange-Correlation Density Functionals ($xc_functional)
- 3.13 User-defined Parameters for DFT Dispersion Correction ($empirical_dispersion)
- 3.14 Addition of External Charges ($external_charges)
- 3.15 Applying a Multipole Field ($multipole_field)
- 3.16 User-Defined Occupied Guess Orbitals ($occupied and $swap_occupied_virtual)
- 3.17 Polarizable Continuum Solvation Models ($pcm)
- 3.18 SS(V)PE Solvation Modeling ($svp and $svpirf)
- 3.19 User-Defined van der Waals Radii ($van_der_waals)
- 3.20 Effective Fragment Potential calculations ($efp_fragments and $efp_params)
- 3.21 Natural Bond Orbital Package ($nbo)
- 3.22 Orbitals, Densities and ESPs on a Mesh ($plots)
- 3.23 Intracules ($intracule)
- 3.24 Geometry Optimization with General Constraints ($opt)
- 3.25 Isotopic Substitutions ($isotopes)
- 3.26 Multiple Jobs in a Single File: Q-Chem Batch Job Files
- 3.27 Q-Chem Output File
- 4 Self-Consistent Field Ground State Methods
- 4.1 Introduction
- 4.2 Hartree–Fock Calculations
- 4.3 Density Functional Theory
- 4.4 SCF Initial Guess
- 4.5 Converging SCF Calculations
- 4.6 Large Molecules and Linear Scaling Methods
- 4.7 Dual-Basis Self-Consistent Field Calculations
- 4.8 Hartree-Fock and Density-Functional Perturbative Corrections
- 4.9 Constrained Density Functional Theory (CDFT)
- 4.10 Configuration Interaction with Constrained Density Functional Theory (CDFT-CI)
- 4.11 Unconventional SCF Calculations
- 4.12 SCF Metadynamics
- 4.13 Ground State Method Summary
- 5 Wavefunction-Based Correlation Methods
- 5.1 Introduction
- 5.2 Møller-Plesset Perturbation Theory
- 5.3 Exact MP2 Methods
- 5.4 Local MP2 Methods
- 5.5 Auxiliary Basis Set (Resolution-of-Identity) MP2 Methods
- 5.6 Short-Range Correlation Methods
- 5.7 Coupled-Cluster Methods
- 5.8 Non-iterative Corrections to Coupled Cluster Energies
- 5.9 Coupled Cluster Active Space Methods
- 5.10 Frozen Natural Orbitals in CCD, CCSD, OD, QCCD, and QCISD Calculations
- 5.11 Non-Hartree-Fock Orbitals in Correlated Calculations
- 5.12 Analytic Gradients and Properties for Coupled-Cluster Methods
- 5.13 Memory Options and Parallelization of Coupled-Cluster Calculations
- 5.14 Simplified Coupled-Cluster Methods Based on a Perfect-Pairing Active Space
- 5.15 Geminal Models
- 6 Open-Shell and Excited-State Methods
- 6.1 General Excited-State Features
- 6.2 Non-Correlated Wavefunction Methods
- 6.3 Time-Dependent Density Functional Theory (TDDFT)
- 6.4 Maximum Overlap Method (MOM) for SCF Excited States
- 6.5 Restricted Open-Shell Kohn-Sham Method for
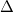 -SCF Calculations of Excited States
-SCF Calculations of Excited States
- 6.6 Correlated Excited State Methods: the CIS(D) Family
- 6.7 Coupled-Cluster Excited-State and Open-Shell Methods
- 6.8 Correlated Excited State Methods: ADC(
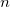 ) Family
) Family
- 6.9 Restricted active space spin-flip (RAS-SF) and configuration interaction (RAS-CI) methods
- 6.10 How to Compute Ionization Energies of Core Electrons and Excited States Involving Excitations of Core Electrons
- 6.11 Visualization of Excited States
- 7 Basis Sets
- 8 Effective Core Potentials
- 9 Molecular Geometry Critical Points and ab Initio Molecular Dynamics
- 9.1 Equilibrium Geometries and Transition Structures
- 9.2 Constrained Optimization
- 9.3 Potential Energy Scans
- 9.4 Minimum-Energy Crossing Points
- 9.5 Intrinsic Reaction Coordinates
- 9.6 Freezing String Method
- 9.7 Hessian-Free Transition State Search
- 9.8 Improved Dimer Method
- 9.9 Ab initio Molecular Dynamics
- 9.10 Ab initio Path Integrals
- 9.11 The EFEI Method
- 10 Molecular Properties and Analysis
- 10.1 Introduction
- 10.2 Wavefunction Analysis
- 10.3 Nonadiabatic couplings
- 10.4 Interface to the NBO Package
- 10.5 Orbital Localization
- 10.6 Visualizing and Plotting Orbitals and Densities
- 10.7 Electrostatic Potentials
- 10.8 Spin and Charge Densities at the Nuclei
- 10.9 Atoms in Molecules
- 10.10 Distributed Multipole Analysis
- 10.11 Intracules
- 10.12 Vibrational Analysis
- 10.13 Anharmonic Vibrational Frequencies
- 10.14 NMR Shielding Tensors
- 10.15 Linear-Scaling NMR Chemical Shifts: GIAO-HF and GIAO-DFT
- 10.16 Indirect Nuclear Spin–Spin Coupling Constants
- 10.17 Linear–Scaling Computation of Electric Properties
- 10.18 Electronic Couplings for Electron Transfer and Energy Transfer
- 10.19 Calculating the Population of Effectively Unpaired (“odd”) Electrons with DFT
- 10.20 Quantum Transport Properties via the Landauer Approximation
- 11 Molecules in Complex Environments: Solvent Models, QM/MM and QM/EFP Features, Density Embedding
- 12 Methods Based on Absolutely-Localized Molecular Orbitals
- 12.1 Introduction
- 12.2 Specifying Fragments in the $molecule Section
- 12.3 FRAGMO Initial Guess for SCF Methods
- 12.4 Locally-Projected SCF Methods
- 12.5 Energy Decomposition and Charge-Transfer Analysis
- 12.6 Job Control for Locally-Projected SCF Methods
- 12.7 The Explicit Polarization (XPol) Method
- 12.8 Symmetry-Adapted Perturbation Theory (SAPT)
- 12.9 The XPol+SAPT (XSAPT) Method
- 12.10 The Many-Body Expansion Method
- A Geometry Optimization with Q-Chem
- B AOINTS
- B.1 Introduction
- B.2 Historical Perspective
- B.3 AOINTS: Calculating ERIs with Q-Chem
- B.4 Shell-Pair Data
- B.5 Shell-Quartets and Integral Classes
- B.6 Fundamental ERI
- B.7 Angular Momentum Problem
- B.8 Contraction Problem
- B.9 Quadratic Scaling
- B.10 Algorithm Selection
- B.11 More Efficient Hartree–Fock Gradient and Hessian Evaluations
- B.12 User-Controllable Variables
- C Q-Chem Quick Reference
- D References and Further Reading