5.1 Introduction
The Hartree-Fock procedure, while often qualitatively correct, is frequently quantitatively deficient. The deficiency is due to the underlying assumption of the Hartree-Fock approximation: that electrons move independently within molecular orbitals subject to an averaged field imposed by the remaining electrons. The error that this introduces is called the correlation energy and a wide variety of procedures exist for estimating its magnitude. The purpose of this Chapter is to introduce the main wavefunction-based methods available in Q-Chem to describe electron correlation.
Wavefunction-based electron correlation methods concentrate on the design of corrections to the wavefunction beyond the mean-field Hartree-Fock description. This is to be contrasted with the density functional theory methods discussed in the previous Chapter. While density functional methods yield a description of electronic structure that accounts for electron correlation subject only to the limitations of present-day functionals (which, for example, omit dispersion interactions), DFT cannot be systematically improved if the results are deficient. Wavefunction-based approaches for describing electron correlation [228, 229] offer this main advantage. Their main disadvantage is relatively high computational cost, particularly for the higher-level theories.
There are four broad classes of models for describing electron correlation that are supported within Q-Chem. The first three directly approximate the full time-independent Schrödinger equation. In order of increasing accuracy, and also increasing cost, they are:
Perturbative treatment of pair correlations between electrons, typically capable of recovering 80% or so of the correlation energy in stable molecules.
Self-consistent treatment of pair correlations between electrons (most often based on coupled-cluster theory), capable of recovering on the order of 95% or so of the correlation energy.
Non-iterative corrections for higher than double substitutions, which can account for more than 99% of the correlation energy. They are the basis of many modern methods that are capable of yielding chemical accuracy for ground state reaction energies, as exemplified by the G2 [202] and G3 methods [203].
These methods are discussed in the following subsections.
There is also a fourth class of methods supported in Q-Chem, which have a different objective. These active space methods aim to obtain a balanced description of electron correlation in highly correlated systems, such as diradicals, or along bond-breaking coordinates. Active space methods are discussed in Section 5.9. Finally, equation-of-motion (EOM) methods provide tools for describing open-shell and electronically excited species. Selected configuration interaction (CI) models are also available.
In order to carry out a wavefunction-based electron correlation calculation using Q-Chem, three $rem variables need to be set:
BASIS to specify the basis set (see Chapter 7)
METHOD for treating correlation
N_FROZEN_CORE frozen core electrons (0 default, optionally FC, or
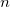 )
)
For wavefunction-based correlation methods, the default option for exchange is Hartree-Fock. If desired, correlated calculations can employ DFT orbitals, which should be set up using a pair of EXCHANGE and CORRELATION keywords. EXCHANGE should be set to a specific DFT method (see Section 5.11).
Additionally, for EOM or CI calculations the number of target states of each type (excited, spin-flipped, ionized, attached, etc.) in each irreducible representation (irrep) should be specified (see Section 6.7.9). The level of correlation of the target EOM states may be different from that used for the reference, and can be specified by EOM_CORR keyword.
The full range of ground and excited state wavefunction-based correlation methods available (i.e. the recognized options to the METHOD keyword) are as follows. Ground-state methods are also a valid option for the CORRELATION keyword.
METHOD
Specifies the level of theory, either DFT or wavefunction-based.
TYPE:
STRING
DEFAULT:
HF
No correlation, Hartree-Fock exchange
OPTIONS:
MP2
RI-MP2
Section 5.5
Local_MP2
Section 5.4
RILMP2
Section 5.5.1
ATTMP2
Section 5.6.1
ATTRIMP2
Section 5.6.1
ZAPT2
A more efficient restricted open-shell MP2 method [230].
MP3
Section 5.2
MP4SDQ
Section 5.2
MP4
Section 5.2
CCD
Section 5.7
CCD(2)
Section 5.8
CCSD
Section 5.7
CCSD(T)
Section 5.8
CCSD(2)
Section 5.8
CCSD(fT)
Section 5.8.3
CCSD(dT)
Section 5.8.3
QCISD
Section 5.7
QCISD(T)
Section 5.8
OD
Section 5.7
OD(T)
Section 5.8
OD(2)
Section 5.8
VOD
Section 5.9
VOD(2)
Section 5.9
QCCD
Section 5.7
QCCD(T)
QCCD(2)
VQCCD
Section 5.9
RECOMMENDATION:
Consult the literature for guidance.