8.1 Introduction
The application of quantum chemical methods to elements in the lower half of the Periodic Table is more difficult than for the lighter atoms. There are two key reasons for this:
the number of electrons in heavy atoms is large
relativistic effects in heavy atoms are often non-negligible
Both of these problems stem from the presence of large numbers of core electrons and, given that such electrons do not play a significant direct role in chemical behavior, it is natural to ask whether it is possible to model their effects in some simpler way. Such enquiries led to the invention of Effective Core Potentials (ECPs) or pseudopotentials. For reviews of relativistic effects in chemistry, see for example Refs. Christiansen:1985,Pyykko:1988,Gordon:1996,Frenking:1996,Cundari:1996,Almlof:1996.
If we seek to replace the core electrons around a given nucleus by a pseudopotential, while affecting the chemistry as little as possible, the pseudopotential should have the same effect on nearby valence electrons as the core electrons. The most obvious effect is the simple electrostatic repulsion between the core and valence regions but the requirement that valence orbitals must be orthogonal to core orbitals introduces additional subtler effects that cannot be neglected.
The most widely used ECPs today are of the form first proposed by Kahn et al. [414] in the 1970s. These model the effects of the core by a one-electron operator 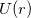 whose matrix elements are simply added to the one-electron Hamiltonian matrix. The ECP operator is given by
whose matrix elements are simply added to the one-electron Hamiltonian matrix. The ECP operator is given by
![\begin{equation} \label{eq800} U(r) = U_ L (r) + \sum \limits _{l=0}^{L-1} { \sum \limits _{m=-l}^{+l} { \left| {Y_{lm} } \right\rangle \left[ {U_ l (r)-U_ L (r)} \right] \left\langle {Y_{lm} } \right|} } \end{equation}](images/img-0949.png) |
(8.1) |
where the 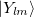 are spherical harmonic projectors and the
are spherical harmonic projectors and the 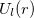 are linear combinations of Gaussians, multiplied by
are linear combinations of Gaussians, multiplied by 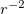 ,
, 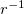 or
or 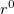 . In addition,
. In addition, 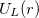 contains a Coulombic term
contains a Coulombic term 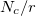 , where
, where 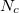 is the number of core electrons.
is the number of core electrons.
One of the key issues in the development of pseudopotentials is the definition of the “core”. So-called “large-core” ECPs include all shells except the outermost one, but “small-core” ECPs include all except the outermost two shells. Although the small-core ECPs are more expensive to use (because more electrons are treated explicitly), it is often found that their enhanced accuracy justifies their use.
When an ECP is constructed, it is usually based either on non-relativistic, or quasi-relativistic all-electron calculations. As one might expect, the quasi-relativistic ECPs tend to yield better results than their non-relativistic brethren, especially for atoms beyond the 3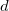 block.
block.