5.13 Memory Options and Parallelization of Coupled-Cluster Calculations
The coupled-cluster suite of methods, which includes ground-state methods mentioned earlier in this Chapter and excited-state methods in the next Chapter, has been parallelized to take advantage of the multi-core architecture. The code is parallelized at the level of the tensor library such that the most time consuming operation, tensor contraction, is performed on different processors (or different cores of the same processor) using shared memory and shared scratch disk space[280].
Parallelization on multiple CPUs or CPU cores is achieved by breaking down tensor operations into batches and running each batch in a separate thread. Because each thread occupies one CPU core entirely, the maximum number of threads must not exceed the total available number of CPU cores. If multiple computations are performed simultaneously, they together should not run more threads than available cores. For example, an eight-core node can accommodate one eight-thread calculation, two four-thread calculations, and so on.
The number of threads to be used in the calculation is specified as a command line option ( -nt nthreads) Here nthreads should be given a positive integer value. If this option is not specified, the job will run in serial mode using single thread only.
Setting the memory limit correctly is also very important for high performance when running larger jobs. To estimate the amount of memory required for coupled-clusters and related calculations, one can use the following formula:
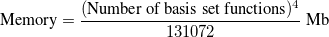 |
(5.41) |
If the new code (CCMAN2) is used and the calculation is based on a RHF reference, the amount of memory needed is a half of that given by the formula. In addition, if gradients are calculated, the amount should be multiplied by two. Because the size of data increases steeply with the size of the molecule computed, both CCMAN and CCMAN2 are able to use disk space to supplement physical RAM if so required. The strategies of memory management in older CCMAN and newer CCMAN2 slightly differ, and that should be taken into account when specifying memory related keywords in the input file.
The MEM_STATIC keyword specifies the amount of memory in megabytes to be made available to routines that run prior to coupled-clusters calculations: Hartree-Fock and electronic repulsion integrals evaluation. A safe recommended value is 500 Mb. The value of MEM_STATIC should rarely exceed 1000–2000 Mb even for relatively large jobs.
The memory limit for coupled-clusters calculations is set by CC_MEMORY. When running older CCMAN, its value is used as the recommended amount of memory, and the calculation can in fact use less or run over the limit. If the job is to run exclusively on a node, CC_MEMORY should be given 50% of all RAM. If the calculation runs out of memory, the amount of CC_MEMORY should be reduced forcing CCMAN to use memory saving algorithms.
CCMAN2 uses a different strategy. It allocates the entire amount of RAM given by CC_MEMORY before the calculation and treats that as a strict memory limit. While that significantly improves the stability of larger jobs, it also requires the user to set the correct value of CC_MEMORY to ensure high performance. The default value of approximately 1.5 Gb is not appropriate for large calculations, especially if the node has more resources available. When running CCMAN2 exclusively on a node, CC_MEMORY should be set to 75–80% of the total available RAM.
Note: When running small jobs, using too large CC_MEMORY in CCMAN2 is not recommended because Q-Chem will allocate more resources than needed for the calculation, which will affect other jobs that you may wish to run on the same node.
In addition, the user should verify that the disk and RAM together have enough space by using the above formula. In cases when CC_MEMORY set up is in conflict with the available space on a particular platform, the CC job may segfault at run time. In such cases readjusting the CC_MEMORY value in the input is necessary so as to eliminate the segfaulting.
MEM_STATIC
Sets the memory for individual Fortran program modules
TYPE:
INTEGER
DEFAULT:
240
corresponding to 240 Mb or 12% of MEM_TOTAL
OPTIONS:
User-defined number of megabytes.
RECOMMENDATION:
For direct and semi-direct MP2 calculations, this must exceed OVN + requirements for AO integral evaluation (32–160 Mb). Up to 2000 Mb for large coupled-clusters calculations.
CC_MEMORY
Specifies the maximum size, in Mb, of the buffers for in-core storage of block-tensors in CCMAN and CCMAN2.
TYPE:
INTEGER
DEFAULT:
50% of MEM_TOTAL. If MEM_TOTAL is not set, use 1.5 Gb. A minimum of
192 Mb is hard-coded.
OPTIONS:
Integer number of Mb
RECOMMENDATION:
Larger values can give better I/O performance and are recommended for systems with large memory (add to your .qchemrc file. When running CCMAN2 exclusively on a node, CC_MEMORY should be set to 75–80% of the total available RAM. )