10.14 NMR Shielding Tensors
NMR spectroscopy is a powerful technique to yield important information on molecular systems in chemistry and biochemistry. Since there is no direct relationship between the measured NMR signals and structural properties, the necessity for a reliable method to predict NMR chemical shifts arises. Examples for such assignments are numerous, for example, assignments of solid-state spectra [536, 537]. The implementation within Q-Chem uses gauge-including atomic orbitals (GIAOs) [538, 539, 540] to calculate the NMR chemical shielding tensors. This scheme has been proven to be reliable an accurate for many applications [541].
The shielding tensor,  , is a second-order property depending on the external magnetic field,
, is a second-order property depending on the external magnetic field, 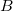 , and the nuclear magnetic spin momentum,
, and the nuclear magnetic spin momentum, 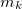 , of nucleus
, of nucleus 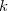 :
:
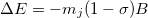 |
(10.62) |
Using analytical derivative techniques to evaluate , the components of this 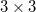 tensor are computed as
tensor are computed as
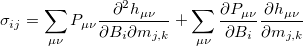 |
(10.63) |
where  and
and 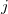 represent are Cartesian components.
represent are Cartesian components.
To solve for the necessary perturbed densities, 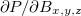 , a new CPSCF method based on a density matrix based formulation [542, 543] is used. This formulation is related to a density matrix based CPSCF (D-CPSCF) formulation employed for the computation of vibrational frequencies [544]. Alternatively, an MO-based CPSCF calculation of shielding tensors can be chosen by the variable MOPROP. Features of the NMR package include:
, a new CPSCF method based on a density matrix based formulation [542, 543] is used. This formulation is related to a density matrix based CPSCF (D-CPSCF) formulation employed for the computation of vibrational frequencies [544]. Alternatively, an MO-based CPSCF calculation of shielding tensors can be chosen by the variable MOPROP. Features of the NMR package include:
Restricted HF-GIAO and KS-DFT-GIAO NMR chemical shifts calculations
LinK/CFMM support to evaluate Coulomb- and exchange-like matrices
Density matrix-based coupled-perturbed SCF (D-CPSCF)
DIIS acceleration
Support of basis sets up to
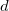 functions
functions Support of LSDA/GGA/Hybrid XC functionals
10.14.1 Job Control
The JOBTYPE must be set to NMR to request the NMR chemical shifts.
D_CPSCF_PERTNUM
Specifies whether to do the perturbations one at a time, or all together.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Perturbed densities to be calculated all together.
1
Perturbed densities to be calculated one at a time.
RECOMMENDATION:
None
D_SCF_CONV_1
Sets the convergence criterion for the level-1 iterations. This preconditions the density for the level-2 calculation, and does not include any two-electron integrals.
TYPE:
INTEGER
DEFAULT:
4
corresponding to a threshold of
.
OPTIONS:
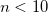
Sets convergence threshold to
.
RECOMMENDATION:
The criterion for level-1 convergence must be less than or equal to the level-2 criterion, otherwise the D-CPSCF will not converge.
D_SCF_CONV_2
Sets the convergence criterion for the level-2 iterations.
TYPE:
INTEGER
DEFAULT:
4
Corresponding to a threshold of
OPTIONS:
Sets convergence threshold to
RECOMMENDATION:
None
D_SCF_MAX_1
Sets the maximum number of level-1 iterations.
TYPE:
INTEGER
DEFAULT:
100
OPTIONS:
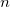
User defined.
RECOMMENDATION:
Use default.
D_SCF_MAX_2
Sets the maximum number of level-2 iterations.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
RECOMMENDATION:
Use default.
D_SCF_DIIS
Specifies the number of matrices to use in the DIIS extrapolation in the D-CPSCF.
TYPE:
INTEGER
DEFAULT:
11
OPTIONS:
RECOMMENDATION:
Use the default.
10.14.2 Using NMR Shielding Constants as an Efficient Probe of Aromaticity
Unambiguous theoretical estimates of degree of aromaticity are still on high demand. The NMR chemical shift methodology offers one unique probe of aromaticity based on one defining characteristics of an aromatic system—its ability to sustain a diatropic ring current. This leads to a response to an imposed external magnetic field with a strong (negative) shielding at the center of the ring. Schleyer and co. have employed this phenomenon to justify a new unique probe of aromaticity [545]. They proposed the computed absolute magnetic shielding at ring centers (unweighted mean of the heavy-atoms ring coordinates) as a new aromaticity criterion, called nucleus-independent chemical shift (NICS). Aromatic rings show strong negative shielding at the ring center (negative NICS), while anti-aromatic systems reveal positive NICS at the ring center. As an example, a typical NICS value for benzene is about -11.5 ppm as estimated with Q-Chem at Hartree-Fock/6-31G* level. The same NICS value for benzene was also reported in Ref. vonSchleyer:1996. The calculated NICS value for furan of 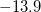 ppm with Q-Chem is about the same as the value reported for furan in Ref. vonSchleyer:1996. Below is one input example of how to the NICS of furan with Q-Chem, using the ghost atom option. The ghost atom is placed at the center of the furan ring, and the basis set assigned to it within the basis mix option must be the basis used for hydrogen atom.
ppm with Q-Chem is about the same as the value reported for furan in Ref. vonSchleyer:1996. Below is one input example of how to the NICS of furan with Q-Chem, using the ghost atom option. The ghost atom is placed at the center of the furan ring, and the basis set assigned to it within the basis mix option must be the basis used for hydrogen atom.
Example 10.227 Calculation of the NMR NICS probe of furan with Hartree-Fock/6-31G* with Q-Chem.
$molecule
0 1
C -0.69480 -0.62270 -0.00550
C 0.72110 -0.63490 0.00300
C 1.11490 0.68300 0.00750
O 0.03140 1.50200 0.00230
C -1.06600 0.70180 -0.00560
H 2.07530 1.17930 0.01410
H 1.37470 -1.49560 0.00550
H -1.36310 -1.47200 -0.01090
H -2.01770 1.21450 -0.01040
GH 0.02132 0.32584 0.00034 ! ghost at the ring center
$end
$rem
JobType NMR
METHOD HF
BASIS mixed
SCF_Algorithm DIIS
PURCAR 111
SEPARATE_JK 0
LIN_K 0
CFMM_ORDER 15
GRAIN 1
CFMM_PRINT 2
CFMMSTAT 1
PRINT_PATH_TIME 1
LINK_MAXSHELL_NUMBER 1
SKIP_SCFMAN 0
IGUESS core ! Core Hamiltonian Guess
SCF_Convergence 7
ITHRSH 10 ! Threshold
IPRINT 23
D_SCF_CONVGUIDE 0 !REM_D_SCF_CONVGUIDE
D_SCF_METRIC 2 !Metric...
D_SCF_STORAGE 50 !REM_D_SCF_STORAGE
D_SCF_RESTART 0 !REM_D_SCF_RESTART
PRINT_PATH_TIME 1
SYM_IGNORE 1
NO_REORIENT 1
$end
$basis
C 1
6-31G*
****
C 2
6-31G*
****
C 3
6-31G*
****
O 4
6-31G*
****
C 5
6-31G*
****
H 6
6-31G*
****
H 7
6-31G*
****
H 8
6-31G*
****
H 9
6-31G*
****
H 10
6-31G*
****