B.2 Historical Perspective
Prior to the 1950s, the most difficult step in the systematic application of Schrödinger wave mechanics to chemistry was the calculation of the notorious two-electron integrals that measure the repulsion between electrons. Boys [748] showed that this step can be made easier (although still time consuming) if Gaussian, rather than Slater, orbitals are used in the basis set. Following the landmark paper of computational chemistry [749] (again due to Boys) programs were constructed that could calculate all the ERIs that arise in the treatment of a general polyatomic molecule with  and
and 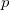 orbitals. However, the programs were painfully slow and could only be applied to the smallest of molecular systems.
orbitals. However, the programs were painfully slow and could only be applied to the smallest of molecular systems.
In 1969, Pople constructed a breakthrough ERI algorithm, a hundred time faster than its predecessors. The algorithm remains the fastest available for its associated integral classes and is now referred to as the Pople-Hehre axis-switch method [750].
Over the two decades following Pople’s initial development, an enormous amount of research effort into the construction of ERIs was documented, which built on Pople’s original success. Essentially, the advances of the newer algorithms could be identified as either better coping with angular momentum (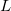 ) or, contraction (
) or, contraction (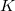 ); each new method increasing the speed and application of quantum mechanics to solving real chemical problems.
); each new method increasing the speed and application of quantum mechanics to solving real chemical problems.
By 1990, another barrier had been reached. The contemporary programs had become sophisticated and both academia and industry had begun to recognize and use the power of ab initio quantum chemistry, but the software was struggling with “dusty deck syndrome” and it had become increasingly difficult for it to keep up with the rapid advances in hardware development. Vector processors, parallel architectures and the advent of the graphical user interface were all demanding radically different approaches to programming and it had become clear that a fresh start, with a clean slate, was both inevitable and desirable. Furthermore, the integral bottleneck had re-emerged in a new guise and the standard programs were now hitting the 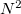 wall. Irrespective of the speed at which ERIs could be computed, the unforgiving fact remained that the number of ERIs required scaled quadratically with the size of the system.
wall. Irrespective of the speed at which ERIs could be computed, the unforgiving fact remained that the number of ERIs required scaled quadratically with the size of the system.
The Q-Chem project was established to tackle this problem and to seek new methods that circumvent the wall. Fundamentally new approaches to integral theory were sought and the ongoing advances that have resulted [551, 220, 221, 751, 752] have now placed Q-Chem firmly at the vanguard of the field. It should be emphasized, however, that the 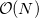 methods that we have developed still require short-range ERIs to treat interactions between nearby electrons, thus the importance of contemporary ERI code remains.
methods that we have developed still require short-range ERIs to treat interactions between nearby electrons, thus the importance of contemporary ERI code remains.
The chronological development and evolution of integral methods can be summarized by considering a time line showing the years in which important new algorithms were first introduced. These are best discussed in terms of the type of ERI or matrix elements that the algorithm can compute efficiently.
1950 |
Boys |
Boys:1950 |
ERIs with low |
1969 |
Pople |
Pople:1978 |
ERIs with low |
1976 |
Dupuis |
Dupuis:1976 |
Integrals with any |
1978 |
McMurchie |
McMurchie:1978 |
Integrals with any |
1982 |
Almlöf |
Almlof:1982 |
Introduction of the direct SCF approach |
1986 |
Obara |
Obara:1986 |
Integrals with any |
1988 |
Head-Gordon |
Head-Gordon:1988b |
Integrals with any |
1991 |
Gill |
Gill:1990,Gill:1991b |
Integrals with any |
1994 |
White |
White:1994a |
J matrix in linear work |
1996 |
Schwegler |
Schwegler:1996a,Schwegler:1996b |
HF exchange matrix in linear work |
1997 |
Challacombe |
Challacombe:1997 |
Fock matrix in linear work |