4.8 Hartree-Fock and Density-Functional Perturbative Corrections
4.8.1 Hartree-Fock Perturbative Correction
An HFPC [208, 209] calculation consists of an iterative HF calculation in a small primary basis followed by a single Fock matrix formation, diagonalization, and energy evaluation in a larger, secondary basis. We denote a conventional HF calculation by HF/basis, and a HFPC calculation by HFPC/primary/secondary. Using a primary basis of 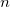 functions, the restricted HF matrix elements for a 2
functions, the restricted HF matrix elements for a 2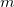 -electron system are
-electron system are
![\begin{equation} F_{\mu \nu } = h_{\mu \nu } + \sum _{\lambda \sigma }^{n} P_{\lambda \sigma } \left[(\mu \nu |\lambda \sigma ) - \frac{1}{2}(\mu \lambda |\nu \sigma )\right] \end{equation}](images/img-0374.png) |
(4.93) |
Solving the Roothaan-Hall equation in the primary basis results in molecular orbitals and an associated density matrix, 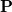 . In an HFPC calculation, is subsequently used to build a new Fock matrix,
. In an HFPC calculation, is subsequently used to build a new Fock matrix, ![$\ensuremath{\mathbf{F}}^{[1]}$](images/img-0376.png) , in a larger secondary basis of
, in a larger secondary basis of 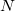 functions
functions
![\begin{equation} F_{ab}^{[1]} = h_{ab} + \sum _{\lambda \sigma }^{n} P_{\lambda \sigma } \left[(ab|\lambda \sigma ) - \frac{1}{2}(a\lambda |b\sigma )\right] \label{eq:F1} \end{equation}](images/img-0377.png) |
(4.94) |
where 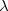 ,
,  indicate primary basis functions and
indicate primary basis functions and  ,
, 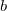 represent secondary basis functions. Diagonalization of yields improved molecular orbitals and an associated density matrix
represent secondary basis functions. Diagonalization of yields improved molecular orbitals and an associated density matrix ![$\ensuremath{\mathbf{P}}^{[1]}$](images/img-0379.png) . The HFPC energy is given by
. The HFPC energy is given by
![\begin{equation} E^\text {HFPC} = \sum _{ab}^ N P^{[1]}_{ab} h_{ab} + \frac{1}{2} \sum _{abcd}^{N} P^{[1]}_{ab}P^{[1]}_{cd} \left[2(ab|cd) - (ac|bd)\right] \label{eq:EHFPC} \end{equation}](images/img-0380.png) |
(4.95) |
where , ,  and
and 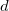 represent secondary basis functions. This differs from the DBHF energy evaluation where
represent secondary basis functions. This differs from the DBHF energy evaluation where ![$\ensuremath{\mathbf{P P}}^{[1]}$](images/img-0382.png) , rather than
, rather than ![$\ensuremath{\mathbf{P}}^{[1]}\ensuremath{\mathbf{P}}^{[1]}$](images/img-0383.png) , is used. The inclusion of contributions that are quadratic in is the key reason for the fact that HFPC is more accurate than DBHF.
, is used. The inclusion of contributions that are quadratic in is the key reason for the fact that HFPC is more accurate than DBHF.
Unlike DBHF, HFPC does not require proper subset/superset basis set combinations and is therefore able to jump between any two basis sets. Benchmark study of HFPC on a large and diverse data set of total and reaction energies show that, for a range of primary/secondary basis set combinations the HFPC scheme can reduce the error of the primary calculation by around two orders of magnitude at a cost of about one third that of the full secondary calculation.
4.8.2 Density Functional Perturbative Correction (Density Functional “Triple Jumping”)
Density Functional Perturbation Theory (DFPC) [210] seeks to combine the low cost of pure calculations using small bases and grids with the high accuracy of hybrid calculations using large bases and grids. Our method is motivated by the dual functional method of Nakajima and Hirao [211] and the dual grid scheme of Tozer et al. [212] We combine these with dual basis ideas to obtain a triple perturbation in the functional, grid and basis directions.
4.8.3 Job Control
HFPC/DFPC calculations are controlled with the following $rem. HFPT turns on the HFPC/DFPC approximation. Note that HFPT_BASIS specifies the secondary basis set.
HFPT
Activates HFPC/DFPC calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Single-point energy only
RECOMMENDATION:
Use Dual-Basis to capture large-basis effects at smaller basis cost. See reference for recommended basis set, functional, and grid pairings.
HFPT_BASIS
Specifies the secondary basis in a HFPC/DFPC calculation.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
None
RECOMMENDATION:
See reference for recommended basis set, functional, and grid pairings.
DFPT_XC_GRID
Specifies the secondary grid in a HFPC/DFPC calculation.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
None
RECOMMENDATION:
See reference for recommended basis set, functional, and grid pairings.
DFPT_EXCHANGE
Specifies the secondary functional in a HFPC/DFPC calculation.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
None
RECOMMENDATION:
See reference for recommended basis set, functional, and grid pairings.
4.8.4 Examples
Example 4.72 Input for a HFPC single-point calculation.
$molecule
0 1
H
H 1 0.75
$end
$rem
JOBTYPE sp
EXCHANGE hf
BASIS cc-pVDZ !primary basis
HFPT_BASIS cc-pVQZ !secondary basis
PURECART 1111 ! set to purecart of the target basis
HFPT true
$end
Example 4.73 Input for a DFPC single-point calculation.
$molecule
0 1
H
H 1 0.75
$end
$rem
JOBTYPE sp
EXCHANGE blyp !primary functional
DFPT_EXCHANGE b3lyp !secondary functional
DFPT_XC_GRID 00075000302 !secondary grid
XC_GRID 0 !primary grid
HFPT_BASIS 6-311++G(3df,3pd) !secondary basis
BASIS 6-311G* !primary basis
PURECART 1111
HFPT true
$end