9.4 Minimum-Energy Crossing Points
Conical intersections are the regions of the potential energy surface characterized by degeneracy between two or more electronic states with the same symmetry. For a two-state intersection, the intersection consists of an 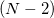 -dimensional hypersurface (the seam space) within which the two states are degenerate, where
-dimensional hypersurface (the seam space) within which the two states are degenerate, where 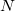 is the number of internal coordinates of the molecule. (The remaining two dimensions for a branching space in which the degeneracy is lifted by any infinitesimal displacement.) Radiationless transitions between the two electronic states are likely to occur in an around a conical seam. The first step in any study of nonadiabatic excited-state dynamics is often an exploration of the geometries and energies of the lowest-energy point within the seam space, which is the so-called minimum-energy crossing point (MECP). In some sense, the MECP is to internal conversion and photochemical processes what the transition state is to single-surface chemical reactions.
is the number of internal coordinates of the molecule. (The remaining two dimensions for a branching space in which the degeneracy is lifted by any infinitesimal displacement.) Radiationless transitions between the two electronic states are likely to occur in an around a conical seam. The first step in any study of nonadiabatic excited-state dynamics is often an exploration of the geometries and energies of the lowest-energy point within the seam space, which is the so-called minimum-energy crossing point (MECP). In some sense, the MECP is to internal conversion and photochemical processes what the transition state is to single-surface chemical reactions.
The two-dimensional branching space between electronic states 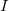 and
and 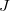 is spanned by a pair of vectors that are usually denoted
is spanned by a pair of vectors that are usually denoted 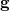 and
and 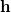 [416]:
[416]:
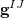 |
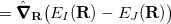 |
(9.2) | ||
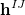 |
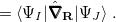 |
(9.3) |
While 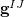 is available analytically for any electronic structure method that has analytic excited-state gradients, analytic implementations of the nonadiabatic coupling vector
is available analytically for any electronic structure method that has analytic excited-state gradients, analytic implementations of the nonadiabatic coupling vector 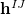 are not routinely available. For this reason, several algorithms have been developed to optimize MECPs without the need to evaluate , and three such algorithms are available in Q-Chem. The first of these is a penalty-constrained optimization algorithm developed by Levine et al. [417], in which the objective function that is optimized to locate the MECP is
are not routinely available. For this reason, several algorithms have been developed to optimize MECPs without the need to evaluate , and three such algorithms are available in Q-Chem. The first of these is a penalty-constrained optimization algorithm developed by Levine et al. [417], in which the objective function that is optimized to locate the MECP is
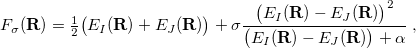 |
(9.4) |
where  is a fixed parameter to avoid singularities and
is a fixed parameter to avoid singularities and  is a Lagrange multiplier for a penalty function meant to drive the energy gap to zero. Optimization of
is a Lagrange multiplier for a penalty function meant to drive the energy gap to zero. Optimization of 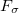 proceeds iteratively for increasingly large values of the parameter . The second MECP optimization algorithm is a simplification of the first one that we call the “direct” method. Here, the gradient of the objective function is
proceeds iteratively for increasingly large values of the parameter . The second MECP optimization algorithm is a simplification of the first one that we call the “direct” method. Here, the gradient of the objective function is
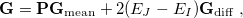 |
(9.5) |
where
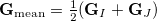 |
(9.6) |
is the mean energy gradient (with 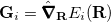 being the nuclear gradient for state
being the nuclear gradient for state  ), and
), and
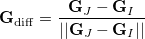 |
(9.7) |
is the normalized difference gradient. Finally,
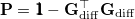 |
(9.8) |
projects the gradient difference direction out of the mean energy gradient in Eq. eq:direct_MECP. The third and final MECP optimization algorithm that is available in Q-Chem is the branching-plane updating method developed by Maeda et al. [418]. This algorithm uses a gradient that is similar to that in Eq. eq:direct_MECP but projects out not just 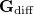 in Eq. eq:MECP_projector but also a second vector that is orthogonal to it.
in Eq. eq:MECP_projector but also a second vector that is orthogonal to it.
None of these three methods requires evaluation of nonadiabatic couplings, and all three can be used to optimize MECPs at the CIS, SF-CIS, TDDFT, SF-TDDFT, and SOS-CIS(D0) levels. The direct algorithm can also be used for EOM-XX-CCSD methods. Note, however, that all linear-response methods (a category that includes CIS, TDDFT, and EOM-XX-CCSD) incorrectly describe the topology of any conical intersection that involves the reference (usually, ground) state. Specifically, it can be shown in such cases that only a one-dimensional branching space is obtained [419]. If conical intersections involving the ground state are to be described correctly, one must use a different reference state, which can be accomplished with spin-flip (SF) methods [420, 421, 422]. In particular, using a high-spin triplet reference state, the ground-state singlet (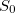 ) appears as an excitation (possibly with a negative excitation energy) alongside the excited-state singlets,
) appears as an excitation (possibly with a negative excitation energy) alongside the excited-state singlets, 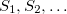 , so there is no topology problem in describing an /
, so there is no topology problem in describing an /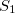 conical intersection. Accurate MECP geometries, as compared to multireference configuration-interaction benchmarks, have been reported [421]. It should be noted, however, that the
conical intersection. Accurate MECP geometries, as compared to multireference configuration-interaction benchmarks, have been reported [421]. It should be noted, however, that the 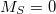 component of the triplet state also shows up as an excitation in SF methods, and while Q-Chem attempts to identify this triplet state automatically (based on a threshold for
component of the triplet state also shows up as an excitation in SF methods, and while Q-Chem attempts to identify this triplet state automatically (based on a threshold for 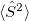 ), severe spin contamination can sometimes hamper one’s ability to distinguish this state from the singlet excited states [421].
), severe spin contamination can sometimes hamper one’s ability to distinguish this state from the singlet excited states [421].
In Q-Chem 4.3, analytic derivative couplings for CIS and TDDFT have been implemented 10.3. Thus, CIS and TDDFT MECP optimizations can use this new feature to reduce the optimization cycles.[422] Note that the $derivative_coupling section is not required in MECP optimization jobs, and $rem variable MECP_METHODS must be set to BRANCHING_PLANE.
9.4.1 Job Control
MECP_OPT
Determines whether we are doing MECP optimizations.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Do MECP optimizations.
FALSE
Don’t do MECP optimizations.
RECOMMENDATION:
None.
MECP_METHODS
Determines which method to be used.
TYPE:
STRING
DEFAULT:
BRANCHING_PLANE
OPTIONS:
BRANCHING_PLANE
Use the branching-plane updating method.
MECP_DIRECT
Use the direct method.
PENALTY_FUNCTION
Use the penalty-constrained method.
RECOMMENDATION:
The direct method is stable for small molecules or molecules with high symmetries, but the branching-plane updating method is more efficient for larger molecules. However, the latter does not work if the two states have different symmetries. If using branching plane updating method, GEOM_OPT_COORDS must be set to 0 in the $rem section (i.e., this algorithm is available in Cartesian coordinates only). The penalty-constrained method converges slowly and is suggested only when the other methods do not work.
MECP_STATE1
Determines the first state for crossing.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
None
OPTIONS:
[
]
find the
means the SCF ground state.
RECOMMENDATION:
MECP_STATE2
Determines the second state for crossing.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
None
OPTIONS:
[
find the
RECOMMENDATION:
CIS_S2_THRESH
Determines whether a state is singlet or triplet in unrestricted calculations.
TYPE:
INTEGER
DEFAULT:
120
OPTIONS:
None
RECOMMENDATION:
If set to 120, the states with
are treated as triplet states, with other states are treated as singlets.
MECP_PROJ_HESS
Determines whether to project out the coupling vector from the Hessian when using branching plane updating method.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
FALSE
RECOMMENDATION:
Use Default.
9.4.2 Examples
The MECP between the S and S
and S states of NO
states of NO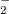 is optimized using the direct method:
is optimized using the direct method:
Example 9.192 MECP optimization for NO using SOS-CIS(D0)
$MOLECULE
-1 1
N1
O2 N1 RNO
O3 N1 RNO O2 AONO
RNO=1.50
AONO=100
$END
$rem
jobtype = opt
method = soscis(d0)
basis = aug-cc-pVDZ
aux_basis = rimp2-aug-cc-pVDZ
purecart = 1111
cis_n_roots = 4
cis_triplets = false
cis_singlets = true
mem_static = 900
mem_total = 1950
mecp_opt true
mecp_state1 [0,2]
mecp_state2 [0,3]
mecp_methods mecp_direct
$end
The MECP between the S and S
and S states of ethylidene is optimized using the branching-plane update method:
states of ethylidene is optimized using the branching-plane update method:
Example 9.193 MECP optimization for ethylidene using SF-TDDFT
$molecule
0 3
C 0.0446266041 -0.2419241370 0.3571573801
C 0.0089051507 0.6727548956 1.4605006396
H 0.9284257388 -0.1459163900 -0.2720952334
H -0.8310326564 -0.1926895078 -0.2885298629
H -0.0092388670 0.9611331703 2.4799363398
H 0.0683140308 -1.2533580302 0.7788470826
$end
$rem
jobtype opt
mecp_opt true
mecp_methods branching_plane
MECP_PROJ_HESS true !project out y vector from the hessian
GEOM_OPT_COORDS 0 !currently only works for Cartesian coordinate
method bhhlyp
spin_flip true
unrestricted true
basis 6-31G(d,p)
cis_n_roots 4
mecp_state1 [0,1]
mecp_state2 [0,2]
CIS_S2_THRESH 120
$end
The MECP between S and S states of twisted-pyramidalized ethylene is optimized using the penalty-constrained method:
Example 9.194 MECP optimization for ethylene using SF-TDDFT
$molecule
0 3
C -0.0158897609 0.0735325545 -0.0595597308
C 0.0124274563 -0.0024687284 1.3156941918
H 0.8578762360 0.1470146857 -0.7105293671
H -0.9364708648 -0.0116961121 -0.6267613144
H 0.7645577838 0.6633816890 1.7625731128
H 0.7407739370 -0.8697640880 1.3285831079
$end
$rem
jobtype opt
mecp_opt true
mecp_methods PENALTY_FUNCTION
method bhhlyp
spin_flip true
unrestricted true
basis 6-31G(d,p)
cis_n_roots 4
mecp_state1 [0,1]
mecp_state2 [0,2]
CIS_S2_THRESH 120
$end
The MECP between 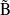
 A and ÃB states of the N
A and ÃB states of the N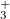 ion using the direct method:
ion using the direct method:
Example 9.195 MECP optimization for N using EOM-EE-CCSD
$MOLECULE
1 1
N1
N2 N1 rNN
N3 N2 rNN N1 aNNN
rNN=1.54
aNNN=50.0
$END
$REM
JOBTYPE OPT
MECP_OPT TRUE
MECP_METHODS mecp_direct
METHOD EOM-CCSD
BASIS 6-31G
ee_singlets [0,2,0,2]
XOPT_STATE_1 [0,2,2]
XOPT_STATE_2 [0,4,1]
ccman2 false
GEOM_OPT_TOL_GRADIENT 30
$END
MECP optimization using analytic derivative coupling:
Example 9.196 MECP between and of ethylidene optimized using BH&HLYP spin-flip TDDFT with analytic derivative coupling
$molecule
0 3
C 0.0446266041 -0.2419241370 0.3571573801
C 0.0089051507 0.6727548956 1.4605006396
H 0.9284257388 -0.1459163900 -0.2720952334
H -0.8310326564 -0.1926895078 -0.2885298629
H -0.0092388670 0.9611331703 2.4799363398
H 0.0683140308 -1.2533580302 0.7788470826
$end
$rem
jobtype opt
mecp_opt true
mecp_methods branching_plane
MECP_PROJ_HESS true
GEOM_OPT_COORDS 0
mecp_state1 [0,1]
mecp_state2 [0,2]
unrestricted true
spin_flip true
cis_n_roots 4
cis_der_couple true
CIS_DER_NUMSTATE 2
set_iter 50
exchange bhhlyp
basis 6-31G(d,p)
symmetry_ignore true
$end