7.8 Basis Set Superposition Error (BSSE)
When calculating binding energies, the energies of the fragments are usually higher than they should be due to the smaller effective basis set used for the individual species. This leads to an overestimate of the binding energy called the basis set superposition error. The effects of this can be corrected for by performing the calculations on the individual species in the presence of the basis set associated with the other species. This requires basis functions to be placed at arbitrary points in space, not just those defined by the nuclear centers. This can be done within Q-Chem by using ghost atoms. These atoms have zero nuclear charge, but can support a user defined basis set. Ghost atom locations are specified in the $molecule section, as for any other atom, and the basis must be specified in a $basis section in the same manner as for a mixed basis.
Example 7.180 A calculation on a water monomer in the presence of the full dimer basis set. The energy will be slightly lower than that without the ghost atom functions due to the greater flexibility of the basis set.
$molecule
0 1
O 1.68668 -0.00318 0.000000
H 1.09686 0.01288 -0.741096
H 1.09686 0.01288 0.741096
Gh -1.45451 0.01190 0.000000
Gh -2.02544 -0.04298 -0.754494
Gh -2.02544 -0.04298 0.754494
$end
$rem
METHOD mp2
BASIS mixed
$end
$basis
O 1
6-31G*
****
H 2
6-31G*
****
H 3
6-31G*
****
O 4
6-31G*
****
H 5
6-31G*
****
H 6
6-31G*
****
$end
Ghosts atoms can also be specified by placing 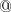 in front of the corresponding atomic symbol in the $molecule section of the input file. If is used to designate the ghost atoms in the system then it is not necessary to use MIXED basis set and include the $basis section in the input.
in front of the corresponding atomic symbol in the $molecule section of the input file. If is used to designate the ghost atoms in the system then it is not necessary to use MIXED basis set and include the $basis section in the input.
Example 7.181 A calculation on ammonia in the presence of the basis set of ammonia borane.
$molecule
0 1
N 0.0000 0.0000 0.7288
H 0.9507 0.0001 1.0947
H -0.4752 -0.8234 1.0947
H -0.4755 0.8233 1.0947
@B 0.0000 0.0000 -0.9379
@H 0.5859 1.0146 -1.2474
@H 0.5857 -1.0147 -1.2474
@H -1.1716 0.0001 -1.2474
$end
$rem
JOBTYPE SP
METHOD B3LYP
BASIS 6-31G(d,p)
PURECART 1112
$end
Finally, there is an alternative approach to counterpoise corrections that is also available in Q-Chem. The powerful Absolutely Localized Molecular Orbital (ALMO) methods can be very conveniently used for the fully automated evaluation of BSSE corrections with associated computational advantages also. This is described in detail, including examples, in Section 12.4.3.