12.9 The XPol+SAPT (XSAPT) Method
12.9.1 Theory
XPol+SAPT, or “XSAPT”, was introduced by Jacobson and Herbert [689, 456] as a low-order-scaling, systematically-improvable method applicable to large systems. The idea is simply to replace the need for empirical parameters in the XPol method with on-the-fly evaluation of exchange-repulsion and dispersion interactions via pairwise-additive SAPT. (This basic idea has spawned several related approaches; see Ref. Lao:2015 for an overview of XSAPT-based methods.) Stated differently, XSAPT uses XPol to evaluate many-body (non-pairwise-additive) polarization effects, but then assumes that dispersion and exchange-repulsion interactions are pairwise additive, and evaluates them via pairwise SAPT0 or SAPT(KS) calculations.
In particular, the zeroth-order Hamiltonian for XSAPT is taken by the the sum of fragment Fock operators defined by the XPol procedure, and the perturbation is the usual SAPT intermolecular perturbation [Eq. eq:pairwise_perturbation] less the intermolecular interactions contained in the XPol fragment Fock operators. A standard SAPT0 correction is then computed for each pair of monomers, using Eq. eq:SAPT_interaction in conjunction with the modified perturbation, to obtain dimer interaction energy 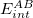 . The total XSAPT energy is then
. The total XSAPT energy is then
![\begin{equation} \label{eq:XSAPT_ total_ E} E^{}_{\rm XSAPT} = \sum _ A \left( \sum _ a \Bigl [ 2\, \epsilon _ a^ A - \mathbf{c}_ a^\dagger ( \mathbf{J}^ A - \tfrac {1}{2}\mathbf{K}^ A)\mathbf{c}_ a\Bigr ] + E_{nuc}^ A + \sum _{B>A} E_{int}^{AB} \right) \; . \end{equation}](images/img-1862.png) |
(12.22) |
In this expression, we have removed the over-counting of two-electron interactions present in Hartree-Fock theory, effectively taking the intrafragment perturbation to first order. The generalization to a Kohn-Sham description of the monomers is straightforward, and is available in Q-Chem.
The inclusion of many-body polarization within the zeroth-order Hamiltonian makes the subsequent SAPT corrections less meaningful in terms of energy decomposition analysis. For instance, the first-order electrostatic correction in XSAPT is not the total electrostatic energy, since the former corrects for errors in the approximate electrostatic treatment at zeroth order (i.e., the electrostatic embedding). The dispersion correction may be less contaminated, since all of the XSAPT modifications to the traditional SAPT perturbation are one-electron operators and therefore the pairwise dispersion correction differs from its traditional SAPT analogue only insofar as the MOs are perturbed by the electrostatic embedding. This should be kept in mind when interpreting the output of an XSAPT calculation, although Lao and Herbert [694, 695] later proposed a many-body energy decomposition scheme for XSAPT that extends traditional SAPT energy decomposition to systems containing more than two monomers. (The aforementioned contamination problems are avoid through pairwise 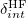 corrections, comparing XSAPT results to traditional SAPT based on gas-phase monomers.) An XSAPT calculation is requested by setting the $rem variables XPOL and SAPT equal to TRUE and also setting XPOL_MPOL_ORDER = CHARGES.
corrections, comparing XSAPT results to traditional SAPT based on gas-phase monomers.) An XSAPT calculation is requested by setting the $rem variables XPOL and SAPT equal to TRUE and also setting XPOL_MPOL_ORDER = CHARGES.
Researchers who use Q-Chem’s XPol+SAPT code are asked to cite Refs. Jacobson:2011 and Herbert:2012. The latter contains a thorough discussion of the theory; a briefer summary can be found in Ref. Jacobson:2013.
Example 12.276 Example showing an XPol+SAPT0 calculation using CHELPG charges and CPHF.
$rem
BASIS CC-PVDZ
METHOD HF
XPOL TRUE
XPOL_MPOL_ORDER CHARGES
XPOL_CHARGE_TYPE QCHELPG
SAPT TRUE
SAPT_CPHF TRUE
SYM_IGNORE TRUE
$end
$molecule
0 1
-- formic acid
0 1
C -1.888896 -0.179692 0.000000
O -1.493280 1.073689 0.000000
O -1.170435 -1.166590 0.000000
H -2.979488 -0.258829 0.000000
H -0.498833 1.107195 0.000000
-- formic acid
0 1
C 1.888896 0.179692 0.000000
O 1.493280 -1.073689 0.000000
O 1.170435 1.166590 0.000000
H 2.979488 0.258829 0.000000
H 0.498833 -1.107195 0.000000
$end
12.9.2 XSAPT(KS)+D
As mentioned above, the dispersion components of the (X)SAPT(KS) interaction energy are not of benchmark quality, even when tuned LRC functionals are employed [713]. The dispersion and exchange-dispersion terms are also the most expensive part of a SAPT0 or SAPT(KS) calculation, scaling as the fourth and fifth powers, respectively, of monomer size, whereas other terms are cubic scaling at worst. Both the efficiency and the accuracy of XSAPT(KS) calculations are thus improved if 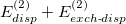 is replaced by an empirical atom–atom dispersion potential. The resulting method is called XSAPT(KS)+D [693, 694, 695]; consult Ref. Lao:2015 for an overview.
is replaced by an empirical atom–atom dispersion potential. The resulting method is called XSAPT(KS)+D [693, 694, 695]; consult Ref. Lao:2015 for an overview.
An XSAPT(KS)+D calculation is requested by setting the $rem variable SAPT_DISP_CORR equal to TRUE. There are three versions of the dispersion potential, a “first generation" (+D1) version [693], second-generation (+D2) version [694], and third-generation (+D3) version [695]; the user can select amongst these using SAPT_DISP_VERSION. Although all three versions exhibit similar performance for total binding energies [695], the +D2 and +D3 potentials were fit directly to ab initio dispersion potentials and do a much better job of reproducing individual energy components, as compared to +D1 [694, 695]. Furthermore, the training set was expanded in the third generation to eliminate the largest errors in +D2 calculations, which tend to be 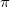 -stacked systems, and +D3 is the recommended dispersion correction.
-stacked systems, and +D3 is the recommended dispersion correction.
As with XPol, the XSAPT and XSAPT(KS)+D methods do not function with a solvation model or with external changes. Currently, only single-point energies are available, and only for closed-shell (restricted) calculations with no frozen orbitals.
Researchers who use XSAPT(KS)+D are asked to cite Ref. Lao:2012b for +D1, Ref. Lao:2013 for +D2, or Ref. Lao:2015 for +D3.
Example 12.277 XSAPT(KS)+D2 calculation of benzene–methane interaction.
$rem
sym_ignore true
exchange gen
basis aug-cc-pvtz
aux_basis rimp2-aug-cc-pvtz
xpol true ! must be set to true for sapt jobs too
xpol_mpol_order charges
xpol_charge_type qchelpg
xpol_omega true
risapt true
sapt_order 2
sapt_basis projected
sapt_disp_corr true
sapt_disp_version 2 ! +D2
thresh 14
scf_convergence 7
basis_lin_dep_thresh 12
lrc_dft true
$end
$xc_functional
x wPBE 1.0
c PBE 1.0
$end
$lrc_omega
275
450
$end
$molecule
0 1
-- benzene
0 1
C 1.3932178 0.0362913 -0.6332803
C 0.7280364 -1.1884015 -0.6333017
C -0.6651797 -1.2247077 -0.6332803
C -1.3932041 -0.0362972 -0.6333017
C -0.7280381 1.1884163 -0.6332803
C 0.6651677 1.2246987 -0.6333017
H 2.4742737 0.0644484 -0.6317240
H 1.2929588 -2.1105409 -0.6317401
H -1.1813229 -2.1750081 -0.6317240
H -2.4742614 -0.0644647 -0.6317401
H -1.2929508 2.1105596 -0.6317240
H 1.1813026 2.1750056 -0.6317401
-- methane
0 1
C 0.0000000 0.0000000 3.0826195
H 0.5868776 0.8381742 3.4463772
H -1.0193189 0.0891638 3.4463772
H 0.0000000 0.0000000 1.9966697
H 0.4324413 -0.9273380 3.4463772
$end
For energy decomposition analysis using, e.g., XSAPT(KS)+D3, three individual calculations must be performed: XSAPT(KS)+D3, SAPT(KS)+D3, and SAPT0, as demonstrated in the example below. The electrostatic, exchange, and dispersion energies can be obtained in the SAPT(KS)+D3 calculation by searching the keywords “E1_elst”, “E1_exch”, and “Empirical E2_disp + E2_exch-disp”, respectively. The induction energy includes three parts. First, there is 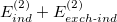 (“E2_ind” and “E2_exch-ind”), obtained from the SAPT(KS)+D3 calculation. The second part is the total energy difference between XSAPT(KS)+D3 and SAPT(KS)+D3, and the total energy can be obtained by searching for “SAPT corrected total energy” in both calculations. This second part provides the many-body polarization and approximate infinite-order response correction. The final part of the energy decomposition accounts for higher-order polarization effects that can be approximated by searching the SAPT0 calculation for the keyword “dSCF” (
(“E2_ind” and “E2_exch-ind”), obtained from the SAPT(KS)+D3 calculation. The second part is the total energy difference between XSAPT(KS)+D3 and SAPT(KS)+D3, and the total energy can be obtained by searching for “SAPT corrected total energy” in both calculations. This second part provides the many-body polarization and approximate infinite-order response correction. The final part of the energy decomposition accounts for higher-order polarization effects that can be approximated by searching the SAPT0 calculation for the keyword “dSCF” (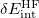 correction). The total binding energy is the sum of electrostatic, exchange, induction, and dispersion components obtained above.
correction). The total binding energy is the sum of electrostatic, exchange, induction, and dispersion components obtained above.
Example 12.278 The XSAPT(KS)+D3 energy-decomposition analysis for water dimer, which requires three individual calculations.
$comment
(1) XSAPT(KS)+D3
$end
$rem
sym_ignore true
exchange gen
basis cc-pvdz
aux_basis rimp2-cc-pvdz
xpol true ! must be set to true for sapt jobs too
xpol_mpol_order charges ! gas or charges
xpol_charge_type qchelpg ! qlowdin,qmulliken,qchelpg
xpol_omega true
xpol_print 3
sapt_print 3
risapt true
sapt_order 2 ! can be set to 1, ELST or RSPT
sapt_basis projected ! monomer, dimer (if only 2 monomers), or projected
sapt_disp_corr true
sapt_disp_version 3
mem_total 4000
mem_static 2000
ao2mo_disk 35000
scf_convergence 7
thresh 12
lrc_dft true
$end
$xc_functional
x wPBE 1.0
c PBE 1.0
$end
$lrc_omega
500
500
$end
$molecule
0 1
--
0 1
O -1.551007 -0.114520 0.000000
H -1.934259 0.762503 0.000000
H -0.599677 0.040712 0.000000
--
0 1
O 1.350625 0.111469 0.000000
H 1.680398 -0.373741 -0.758561
H 1.680398 -0.373741 0.758561
$end
@@@
$comment
(2) SAPT(KS)+D3
$end
$rem
sym_ignore true
exchange gen
basis cc-pvdz
aux_basis rimp2-cc-pvdz
xpol true ! must be set to true for sapt jobs too
xpol_mpol_order gas ! gas or charges
xpol_omega true
xpol_print 3
sapt_print 3
risapt true
sapt_order 2 ! can be set to 1, ELST or RSPT
sapt_basis projected ! monomer, dimer (if only 2 monomers), or projected
sapt_disp_corr true
sapt_disp_version 3
mem_total 4000
mem_static 2000
ao2mo_disk 35000
scf_convergence 7
thresh 12
lrc_dft true
$end
$xc_functional
x wPBE 1.0
c PBE 1.0
$end
$lrc_omega
500
500
$end
$molecule
0 1
--
0 1
O -1.551007 -0.114520 0.000000
H -1.934259 0.762503 0.000000
H -0.599677 0.040712 0.000000
--
0 1
O 1.350625 0.111469 0.000000
H 1.680398 -0.373741 -0.758561
H 1.680398 -0.373741 0.758561
$end
@@@
$comment
(3) SAPT0
$end
$rem
sym_ignore true
exchange hf
basis cc-pvdz
aux_basis rimp2-cc-pvdz
xpol true ! must be set to true for sapt jobs too
xpol_mpol_order gas ! gas or charges
xpol_print 3
sapt_print 3
risapt true
sapt_order 2 ! can be set to 1, ELST or RSPT
sapt_basis dimer ! monomer, dimer (if only 2 monomers), or projected
sapt_cphf true
sapt_dscf true
mem_total 4000
mem_static 2000
ao2mo_disk 35000
scf_convergence 7
thresh 12
$end
$molecule
0 1
--
0 1
O -1.551007 -0.114520 0.000000
H -1.934259 0.762503 0.000000
H -0.599677 0.040712 0.000000
--
0 1
O 1.350625 0.111469 0.000000
H 1.680398 -0.373741 -0.758561
H 1.680398 -0.373741 0.758561
$end