4.7 Dual-Basis Self-Consistent Field Calculations
The dual-basis approximation [196, 197, 198, 199, 200, 201] to self-consistent field (HF or DFT) energies provides an efficient means for obtaining large basis set effects at vastly less cost than a full SCF calculation in a large basis set. First, a full SCF calculation is performed in a chosen small basis (specified by BASIS2). Second, a single SCF-like step in the larger, target basis (specified, as usual, by BASIS) is used to perturbatively approximate the large basis energy. This correction amounts to a first-order approximation in the change in density matrix, after the single large-basis step:
![\begin{equation} E_{\mathrm{total}} = E_{\mathrm{small\; basis}} + \mathrm{Tr}[(\Delta \ensuremath{\mathbf{P}})\cdot \textbf{F}]_{\mathrm{large\; basis}} \end{equation}](images/img-0367.png) |
(4.92) |
where 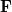 (in the large basis) is built from the converged (small basis) density matrix. Thus, only a single Fock build is required in the large basis set. Currently, HF and DFT energies (SP) as well as analytic first derivatives (FORCE or OPT) are available. [Note: As of version 4.0, first derivatives of unrestricted dual-basis DFT energies—though correct—require a code-efficiency fix. We do not recommend use of these derivatives until this improvement has been made.]
(in the large basis) is built from the converged (small basis) density matrix. Thus, only a single Fock build is required in the large basis set. Currently, HF and DFT energies (SP) as well as analytic first derivatives (FORCE or OPT) are available. [Note: As of version 4.0, first derivatives of unrestricted dual-basis DFT energies—though correct—require a code-efficiency fix. We do not recommend use of these derivatives until this improvement has been made.]
Across the G3 set [202, 203, 204] of 223 molecules, using cc-pVQZ, dual-basis errors for B3LYP are 0.04 kcal/mol (energy) and 0.03 kcal/mol (atomization energy per bond) and are at least an order of magnitude less than using a smaller basis set alone. These errors are obtained at roughly an order of magnitude savings in cost, relative to the full, target-basis calculation.
4.7.1 Dual-Basis MP2
The dual-basis approximation can also be used for the reference energy of a correlated second-order Møller-Plesset (MP2) calculation [197, 201]. When activated, the dual-basis HF energy is first calculated as described above; subsequently, the MO coefficients and orbital energies are used to calculate the correlation energy in the large basis. This technique is particularly effective for RI-MP2 calculations (see Section 5.5), in which the cost of the underlying SCF calculation often dominates.
Furthermore, efficient analytic gradients of the DB-RI-MP2 energy have been developed [199] and added to Q-Chem. These gradients allow for the optimization of molecular structures with RI-MP2 near the basis set limit. Typical computational savings are on the order of 50% (aug-cc-pVDZ) to 71% (aug-cc-pVTZ). Resulting dual-basis errors are only 0.001 in molecular structures and are, again, significantly less than use of a smaller basis set alone.
4.7.2 Basis Set Pairings
We recommend using basis pairings in which the small basis set is a proper subset of the target basis (6-31G into 6-31G*, for example). They not only produce more accurate results; they also lead to more efficient integral screening in both energies and gradients. Subsets for many standard basis sets (including Dunning-style cc-pV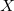 Z basis sets and their augmented analogs) have been developed and thoroughly tested for these purposes. A summary of the pairings is provided in Table 4.4; details of these truncations are provided in Figure 4.1.
Z basis sets and their augmented analogs) have been developed and thoroughly tested for these purposes. A summary of the pairings is provided in Table 4.4; details of these truncations are provided in Figure 4.1.
A new pairing for 6-31G*-type calculations is also available. The 6-4G subset (named r64G in Q-Chem) is a subset by primitive functions and provides a smaller, faster alternative for this basis set regime [200]. A case-dependent switch in the projection code (still OVPROJECTION) properly handles 6-4G. For DB-HF, the calculations proceed as described above. For DB-DFT, empirical scaling factors (see Ref. Steele:2007 for details) are applied to the dual-basis correction. This scaling is handled automatically by the code and prints accordingly.
As of Q-Chem version 3.2, the basis set projection code has also been adapted to properly account for linear dependence [201], which can often be problematic for large, augmented (aug-cc-pVTZ, etc..) basis set calculations. The same standard keyword (LINDEPTHRESH) is utilized for linear dependence in the projection code. Because of the scheme utilized to account for linear dependence, only proper-subset pairings are now allowed.
Like single-basis calculations, user-specified general or mixed basis sets may be employed (see Chapter 7) with dual-basis calculations. The target basis specification occurs in the standard $basis section. The smaller, secondary basis is placed in a similar $basis2 section; the syntax within this section is the same as the syntax for $basis. General and mixed small basis sets are activated by BASIS2=BASIS2_GEN and BASIS2=BASIS2_MIXED, respectively.
BASIS |
BASIS2 |
cc-pVTZ |
rcc-pVTZ |
cc-pVQZ |
rcc-pVQZ |
aug-cc-pVDZ |
racc-pVDZ |
aug-cc-pVTZ |
racc-pVTZ |
aug-cc-pVQZ |
racc-pVQZ |
6-31G* |
r64G, 6-31G |
6-31G** |
r64G, 6-31G |
6-31++G** |
6-31G* |
6-311++G(3df,3pd) |
6-311G*, 6-311+G* |
![\[ \begin{array}{cccccc} $H-He:$ & \scriptsize \begin{array}{ccc} s \\ & p \\ s & & d \\ & p \\ s \\ \end{array} & \scriptsize \begin{array}{ccc} s \\ & p \\ s & & - \\ & - \\ s \\ \end{array} & $Li-Ne:$ & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & d \\ & p & & f\\ s & & d \\ & p \\ s \\ \end{array} & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & d \\ & p & & -\\ s & & d \\ & p \\ s \\ \end{array}\\ & $cc-pVTZ$ & $rcc-pVTZ$ & & $cc-pVTZ$ & $rcc-pVTZ$ \\ \end{array}\\ \]](images/img-0368.png) |
![\[ \begin{array}{cccccc} $H-He:$ & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & d \\ & p & & f\\ s & & d \\ & p \\ s \\ \end{array} & \scriptsize \begin{array}{cccc} s \\ & - \\ s & & - \\ & p & & -\\ s & & - \\ & - \\ s \\ \end{array} & $Li-Ne:$ & \scriptsize \begin{array}{ccccc} s \\ & p \\ s & & d \\ & p & & f\\ s & & d & & g\\ & p & & f\\ s & & d \\ & p \\ s \\ \end{array} & \scriptsize \begin{array}{ccccc} s \\ & p \\ s & & d \\ & p & & -\\ s & & d & & -\\ & p & & -\\ s & & d \\ & p \\ s \\ \end{array}\\ & $cc-pVQZ$ & $rcc-pVQZ$ & & $cc-pVQZ$ & $rcc-pVQZ$ \\ \end{array}\\ \]](images/img-0369.png) |
![\[ \begin{array}{cccccc} $H-He:$ & \scriptsize \begin{array}{ccc} s \\ & p \\ s & \\ & p \\ s’ & \\ \end{array} & \scriptsize \begin{array}{ccc} s \\ & p \\ s & \\ & - \\ s’ & \\ \end{array} & $Li-Ne:$ & \scriptsize \begin{array}{ccc} s \\ & p \\ s & & d \\ & p \\ s & & d’ \\ & p’ \\ s’ \end{array} & \scriptsize \begin{array}{ccc} s \\ & p \\ s & & d \\ & p \\ s & & - \\ & p’ \\ s’ \end{array}\\ & $aug-cc-pVDZ$ & $racc-pVDZ$ & & $aug-cc-pVDZ$ & $racc-pVDZ$ \\ \end{array} \\ \]](images/img-0370.png) |
![\[ \begin{array}{cccccc} $H-He:$ & \scriptsize \begin{array}{ccc} s \\ & p \\ s & & d \\ & p \\ s & & d’ \\ & p’ \\ s’ \end{array} & \scriptsize \begin{array}{ccc} s \\ & p \\ s & & - \\ & p \\ s & & - \\ & - \\ s’ \end{array} & $Li-Ne:$ & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & d \\ & p & & f\\ s & & d \\ & p & & f’\\ s & & d’ \\ & p’ \\ s’ \end{array} & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & d \\ & p & & -\\ s & & d \\ & p & & -\\ s & & - \\ & p’ \\ s’ \end{array}\\ & $aug-cc-pVTZ$ & $racc-pVTZ$ & & $aug-cc-pVTZ$ & $racc-pVTZ$ \\ \end{array} \\ \]](images/img-0371.png) |
![\[ \begin{array}{cccccc} $H-He:$ & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & d \\ & p & & f\\ s & & d \\ & p & & f’\\ s & & d’ \\ & p’ \\ s’ \end{array} & \scriptsize \begin{array}{cccc} s \\ & p \\ s & & - \\ & p & & -\\ s & & - \\ & p & & -\\ s & & - \\ & - \\ s’ \end{array} & $Li-Ne:$ & \scriptsize \begin{array}{ccccc} s \\ & p \\ s & & d \\ & p & & f\\ s & & d & & g\\ & p & & f\\ s & & d & & g’\\ & p & & f’\\ s & & d’ \\ & p’ \\ s’ \end{array} & \scriptsize \begin{array}{ccccc} s \\ & p \\ s & & d \\ & p & & -\\ s & & d & & -\\ & p & & -\\ s & & d & & -\\ & p & & -\\ s & & - \\ & p’ \\ s’ \end{array}\\ & $aug-cc-pVQZ$ & $racc-pVQZ$ & & $aug-cc-pVQZ$ & $racc-pVQZ$ \\ \end{array} \]](images/img-0372.png) |
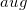 (diffuse) functions. Dashes indicate eliminated functions, relative to the paired standard basis set. In each case, the truncations for hydrogen and heavy atoms are shown, along with the nomenclature used in Q-Chem.
(diffuse) functions. Dashes indicate eliminated functions, relative to the paired standard basis set. In each case, the truncations for hydrogen and heavy atoms are shown, along with the nomenclature used in Q-Chem.4.7.3 Job Control
Dual-Basis calculations are controlled with the following $rem. DUAL_BASIS_ENERGY turns on the Dual-Basis approximation. Note that use of BASIS2 without DUAL_BASIS_ENERGY only uses basis set projection to generate the initial guess and does not invoke the Dual-Basis approximation (see Section 4.4.5). OVPROJECTION is used as the default projection mechanism for Dual-Basis calculations; it is not recommended that this be changed. Specification of SCF variables (e.g., THRESH) will apply to calculations in both basis sets.
DUAL_BASIS_ENERGY
Activates dual-basis SCF (HF or DFT) energy correction.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Analytic first derivative available for HF and DFT (see JOBTYPE)
Can be used in conjunction with MP2 or RI-MP2
See BASIS, BASIS2, BASISPROJTYPE
RECOMMENDATION:
Use Dual-Basis to capture large-basis effects at smaller basis cost. Particularly useful with RI-MP2, in which HF often dominates. Use only proper subsets for small-basis calculation.
4.7.4 Examples
Example 4.67 Input for a Dual-Basis B3LYP single-point calculation.
$molecule
0 1
H
H 1 0.75
$end
$rem
JOBTYPE sp
METHOD b3lyp
BASIS 6-311++G(3df,3pd)
BASIS2 6-311G*
DUAL_BASIS_ENERGY true
$end
Example 4.68 Input for a Dual-Basis B3LYP single-point calculation with a minimal 6-4G small basis.
$molecule
0 1
H
H 1 0.75
$end
$rem
JOBTYPE sp
METHOD b3lyp
BASIS 6-31G*
BASIS2 r64G
DUAL_BASIS_ENERGY true
$end
Example 4.69 Input for a Dual-Basis RI-MP2 single-point calculation.
$molecule
0 1
H
H 1 0.75
$end
$rem
JOBTYPE sp
METHOD rimp2
AUX_BASIS rimp2-cc-pVQZ
BASIS cc-pVQZ
BASIS2 rcc-pVQZ
DUAL_BASIS_ENERGY true
$end
Example 4.70 Input for a Dual-Basis RI-MP2 geometry optimization.
$molecule
0 1
H
H 1 0.75
$end
$rem
JOBTYPE opt
METHOD rimp2
AUX_BASIS rimp2-aug-cc-pVDZ
BASIS aug-cc-pVDZ
BASIS2 racc-pVDZ
DUAL_BASIS_ENERGY true
$end
Example 4.71 Input for a Dual-Basis RI-MP2 single-point calculation with mixed basis sets.
$molecule
0 1
H
O 1 1.1
H 2 1.1 1 104.5
$end
$rem
JOBTYPE opt
METHOD rimp2
AUX_BASIS aux_mixed
BASIS mixed
BASIS2 basis2_mixed
DUAL_BASIS_ENERGY true
$end
$basis
H 1
cc-pVTZ
****
O 2
aug-cc-pVTZ
****
H 3
cc-pVTZ
****
$end
$basis2
H 1
rcc-pVTZ
****
O 2
racc-pVTZ
****
H 3
rcc-pVTZ
****
$end
$aux_basis
H 1
rimp2-cc-pVTZ
****
O 2
rimp2-aug-cc-pVTZ
****
H 3
rimp2-cc-pVTZ
****
$end
4.7.5 Dual-Basis Dynamics
The ability to compute SCF and MP2 energies and forces at reduced cost makes dual-basis calculations attractive for ab initio molecular dynamics simulations. Dual-basis BOMD has demonstrated [205] savings of 58%, even relative to state-of-the-art, Fock-extrapolated BOMD. Savings are further increased to 71% for dual-basis RI-MP2 dynamics. Notably, these timings outperform estimates of extended-Lagrangian (Car-Parrinello) dynamics, without detrimental energy conservation artifacts that are sometimes observed in the latter [206].
Two algorithmic factors make modest but worthwhile improvements to dual-basis dynamics. First, the iterative, small-basis calculation can benefit from Fock matrix extrapolation [206]. Second, extrapolation of the response equations (the so-called “Z-vector” equations) for nuclear forces further increases efficiency [207] . Both sets of keywords are described in Section 9.9, and the code automatically adjusts to extrapolate in the proper basis set when DUAL_BASIS_ENERGY is activated.