C.1 Q-Chem Text Input Summary
Keyword |
Description |
$molecule |
Contains the molecular coordinate input (input file requisite). |
$rem |
Job specification and customization parameters (input file requisite). |
$end |
Terminates each keyword section. |
$basis |
User-defined basis set information (see Chapter 7). |
$comment |
User comments for inclusion into output file. |
$ecp |
User-defined effective core potentials (see Chapter 8). |
$empirical_dispersion |
User-defined van der Waals parameters for DFT dispersion |
correction. |
|
$external_charges |
External charges and their positions. |
$force_field_params |
Force field parameters for QM/MM calculations (see Section‘11.3). |
$intracule |
Intracule parameters (see Chapter 10). |
$isotopes |
Isotopic substitutions for vibrational calculations (see Chapter 10). |
$localized_diabatization |
Information for mixing together multiple adiabatic states into |
diabatic states (see Chapter 10). |
|
$multipole_field |
Details of a multipole field to apply. |
$nbo |
Natural Bond Orbital package. |
$occupied |
Guess orbitals to be occupied. |
$opt |
Constraint definitions for geometry optimizations. |
$pcm |
Special parameters for polarizable continuum models (see Section |
11.2.3). |
|
$plots |
Generate plotting information over a grid of points (see |
Chapter 10). |
|
$qm_atoms |
Specify the QM region for QM/MM calculations (see Section 11.3). |
$solvent |
Additional parameters and variables for implicit solvent models (see Section 11.2). |
$svp |
Special parameters for the SS(V)PE module. |
$svpirf |
Initial guess for SS(V)PE) module. |
$van_der_waals |
User-defined atomic radii for Langevin dipoles solvation (see |
Chapter 10). |
|
$xc_functional |
Details of user-defined DFT exchange-correlation functionals. |
$cdft |
Special options for the constrained DFT method as implemented. |
$efp_fragments |
Specifies labels and positions of EFP fragments. |
$efp_params |
Contains user-defined parameters for effective fragments. |
$eom_user_guess |
Contains user-defined guess for EOM-CC calculations. |
$complex_ccman |
Contains parameters for complex-scaled and CAP-augmented EOM-CC calculations. |
Note: (1) Users are able to enter keyword sections in any order.
(2) Each keyword section must be terminated with the $end keyword.
(3) Not all keywords have to be entered, but $rem and $molecule are compulsory.
(4) Each keyword section will be described below.
(5) The entire Q-Chem input is case–insensitive.
(6) Multiple jobs are separated by the string @@@ on a single line.
C.1.1 Keyword: $molecule
Four methods are available for inputing geometry information:
Z-matrix (Angstroms and degrees):
$molecule
{Z-matrix}
{blank line, if parameters are being used}
{Z-matrix parameters, if used}
$endCartesian Coordinates (Angstroms):
$molecule
{Cartesian coordinates}
{blank line, if parameter are being used}
{Coordinate parameters, if used}
$endRead from a previous calculation:
$molecule
read
$endRead from a file:
$molecule
read filename
$end
C.1.2 Keyword: $rem
See also the list of $rem variables at the end of this Appendix. The general format is:
$rem REM_VARIABLE VALUE [optional comment] $end
C.1.3 Keyword: $basis
The format for the user–defined basis section is as follows:
where
$basis
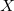
0
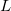
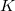
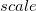
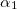
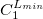
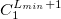

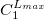
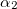
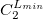
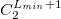
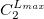


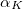
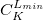
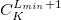
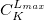
****
$end
|
Atomic symbol of the atom (atomic number not accepted) |
|
Angular momentum symbol (S, P, SP, D, F, G) |
|
Degree of contraction of the shell (integer) |
|
Scaling to be applied to exponents (default is 1.00) |
|
Gaussian primitive exponent (positive real number) |
|
Contraction coefficient for each angular momentum (non–zero real numbers). |
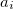
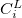
Atoms are terminated with **** and the complete basis set is terminated with the $end keyword terminator. No blank lines can be incorporated within the general basis set input. Note that more than one contraction coefficient per line is one required for compound shells like SP. As with all Q-Chem input deck information, all input is case–insensitive.
C.1.4 Keyword: $comment
Note that the entire input deck is echoed to the output file, thus making the $comment keyword largely redundant.
$comment User comments - copied to output file $end
C.1.5 Keyword: $ecp
$ecp
For each atom that will bear an ECP
Chemical symbol for the atom
ECP name; the value for the ECP; number of core electrons removed
For each ECP component (in the order unprojected, 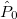 ,
, 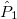 , ,
, , 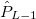
The component name
The number of Gaussians in the component
For each Gaussian in the component
The power of  ; the exponent; the contraction coefficient
; the exponent; the contraction coefficient
****
$end
Note: (1) All of the information in the $ecp block is case–insensitive.
(2) The value may not exceed 4. That is, nothing beyond 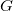 projectors is allowed.
projectors is allowed.
(3) The power of (which includes the Jacobian 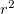 factor) must be 0, 1 or 2.
factor) must be 0, 1 or 2.
C.1.6 Keyword: $empirical_dispersion
$empirical_dispersion S6 S6_value D D_value C6 element_1 C6_value_for_element_1 element_2 C6_value_for_element_2 VDW_RADII element_1 radii_for_element_1 element_2 radii_for_element_2 $end
Note: This section is only for values that the user wants to change from the default values recommended by Grimme.
C.1.7 Keyword: $external_charges
All input should be given in atomic units.
Update: While charges should indeed be listed in atomic units, the units for distances depend on the user input. If the structure is specified in Angstroms (the default), the coordinates for external charges should also be in Angstroms. If the structure is specified in atomic units, the coordinates for external charges should also be in atomic units. (See INPUT_BOHR.)
$external_charges x-coord1 y-coord1 z-coord1 charge1 x-coord2 y-coord2 z-coord2 charge2 $end
C.1.8 Keyword: $intracule
$intracule
$end
int_type
0
Compute
only
1
Compute
only
2
Compute
only
3
Compute
4
Compute
5
Compute
6
Compute
u_points
Number of points, start, end.
v_points
Number of points, start, end.
moments
0–4
Order of moments to be computed (
derivs
0–4
order of derivatives to be computed (
accuracy
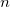
(
specify accuracy of intracule interpolation table (
C.1.9 Keyword: $isotopes
Note that masses should be given in atomic units.
$isotopes
number_extra_loops tp_flag
number_of_atoms [temp pressure]
atom_number1 mass1
atom_number2 mass2
...
$end
C.1.10 Keyword: $multipole_field
Multipole fields are all in atomic units.
$multipole_field field_component1 value1 field_component2 value2 ... $end
C.1.11 Keyword: $nbo
Refer to Chapter 10 and the NBO manual for further information. Note that the NBO $rem variable must be set to ON to initiate the NBO package.
$nbo [ NBO options ] $end
C.1.12 Keyword: $occupied
$occupied 1 2 3 4 ... nalpha 1 2 3 4 ... nbeta $end
C.1.13 Keyword: $opt
Note that units are in Angstroms and degrees. Also see the summary in the next section of this Appendix.
$opt CONSTRAINT stre atom1 atom2 value ... bend atom1 atom2 atom3 value ... outp atom1 atom2 atom3 atom4 value ... tors atom1 atom2 atom3 atom4 value ... linc atom1 atom2 atom3 atom4 value ... linp atom1 atom2 atom3 atom4 value ... ENDCONSTRAINT FIXED atom coordinate_reference ... ENDFIXED DUMMY idum type list_length defining_list ... ENDDUMMY CONNECT atom list_length list ... ENDCONNECT $end
C.1.14 Keyword: $svp
$svp <KEYWORD>=<VALUE>, <KEYWORD>=<VALUE>,... <KEYWORD>=<VALUE> $end
For example, the section may look like this:
$svp
RHOISO=0.001, DIELST=78.39, NPTLEB=110
$end
C.1.15 Keyword: $svpirf
$svpirf
<# point> <x point> <y point> <z point> <charge> <grid weight>
<# point> <x normal> <y normal> <z normal>
$end
C.1.16 Keyword: $plots
$plots
One comment line
Specification of the 3–D mesh of points on 3 lines:
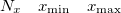
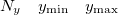
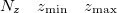
A line with 4 integers indicating how many things to plot:
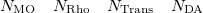
An optional line with the integer list of MO’s to evaluate (only if 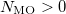 )
)
MO(1) MO(2) MO(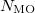 )
)
An optional line with the integer list of densities to evaluate (only if 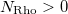 )
)
Rho(1) Rho(2) Rho(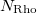 )
)
An optional line with the integer list of transition densities (only if 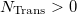 )
)
Trans(1) Trans(2) Trans(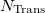 )
)
An optional line with states for detachment/attachment densities (if 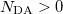 )
)
DA(1) DA(2) DA(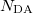 )
)
$end
C.1.17 Keyword: $localized_diabatization
$plots
One comment line.
One line with an an array of adiabatic states to mix together.
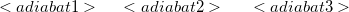
$end
Note: We count adiabatic states such that the first excited state is 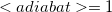 , the fifth is
, the fifth is 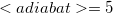 , and so forth.
, and so forth.
C.1.18 Keyword $van_der_waals
Note: all radii are given in angstroms.
$van_der_waals 1 atomic_number VdW_radius $end
(alternative format)
$van_der_waals 2 sequential_atom_number VdW_radius $end
C.1.19 Keyword: $xc_functional
$xc_functional X exchange_symbol coefficient X exchange_symbol coefficient ... C correlation_symbol coefficient C correlation_symbol coefficient ... K coefficient $end