8.4 Pseudopotentials and Density Functional Theory
Q-Chem’s pseudopotential package and DFT package are tightly integrated and facilitate the application of advanced density functionals to molecules containing heavy elements. Any of the local, gradient-corrected and hybrid functionals discussed in Chapter 4 may be used and you may also perform ECP calculations with user-defined hybrid functionals.
In a DFT calculation with pseudopotentials, the exchange-correlation energy is obtained entirely from the non-core electrons. This will be satisfactory if there are no chemically important core-valence effects but may introduce significant errors, particularly if you are using a “large-core” ECP.
Q-Chem’s default quadrature grid is SG-1 (see section 4.3.13) which was originally defined only for the elements up to argon. In Q-Chem 2.0 and above, the SG-1 grid has been extended and it is now defined for all atoms up to, and including, the actinides.
8.4.1 Example
Example 8.185 Optimization of the structure of XeF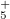 using B3LYP theory and the ECPs of Stevens and collaborators. Note that the BASIS keyword has been omitted and, therefore, the matching SBKJC orbital basis set will be used.
using B3LYP theory and the ECPs of Stevens and collaborators. Note that the BASIS keyword has been omitted and, therefore, the matching SBKJC orbital basis set will be used.
$molecule
1 1
Xe
F1 Xe r1
F2 Xe r2 F1 a
F3 Xe r2 F1 a F2 90
F4 Xe r2 F1 a F3 90
F5 Xe r2 F1 a F4 90
r1 = 2.07
r2 = 2.05
a = 80.0
$end
$rem
JOBTYP opt
METHOD b3lyp
ECP sbkjc
$end