6.6 Correlated Excited State Methods: the CIS(D) Family
CIS(D) [345, 346] is a simple size-consistent doubles correction to CIS which has a computational cost scaling as the fifth power of the basis set for each excited state. In this sense, CIS(D) can be considered as an excited state analog of the ground state MP2 method. CIS(D) yields useful improvements in the accuracy of excitation energies relative to CIS, and yet can be applied to relatively large molecules using Q-Chem’s efficient integrals transformation package. In addition, as in the case of MP2 method, the efficiency can be significantly improved through the use of the auxiliary basis expansions (Section 5.5) [347].
6.6.1 CIS(D) Theory
The CIS(D) excited state procedure is a second-order perturbative approximation to the computationally expensive CCSD, based on a single excitation configuration interaction (CIS) reference. The coupled-cluster wavefunction, truncated at single and double excitations, is the exponential of the single and double substitution operators acting on the Hartree-Fock determinant:
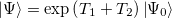 |
(6.15) |
Determination of the singles and doubles amplitudes requires solving the two equations
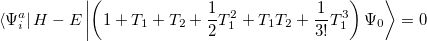 |
(6.16) |
and
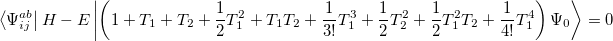 |
(6.17) |
which lead to the CCSD excited state equations. These can be written
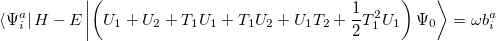 |
(6.18) |
and
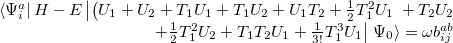 |
(6.19) |
This is an eigenvalue equation 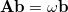 for the transition amplitudes (
for the transition amplitudes (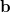 vectors), which are also contained in the
vectors), which are also contained in the 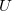 operators.
operators.
The second-order approximation to the CCSD eigenvalue equation yields a second-order contribution to the excitation energy which can be written in the form
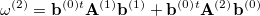 |
(6.20) |
or in the alternative form
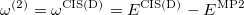 |
(6.21) |
where
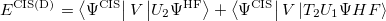 |
(6.22) |
and
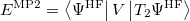 |
(6.23) |
The output of a CIS(D) calculation contains useful information beyond the CIS(D) corrected excitation energies themselves. The stability of the CIS(D) energies is tested by evaluating a diagnostic, termed the “theta diagnostic” [348]. The theta diagnostic calculates a mixing angle that measures the extent to which electron correlation causes each pair of calculated CIS states to couple. Clearly the most extreme case would be a mixing angle of 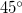 , which would indicate breakdown of the validity of the initial CIS states and any subsequent corrections. On the other hand, small mixing angles on the order of only a degree or so are an indication that the calculated results are reliable. The code can report the largest mixing angle for each state to all others that have been calculated.
, which would indicate breakdown of the validity of the initial CIS states and any subsequent corrections. On the other hand, small mixing angles on the order of only a degree or so are an indication that the calculated results are reliable. The code can report the largest mixing angle for each state to all others that have been calculated.
6.6.2 Resolution of the Identity CIS(D) Methods
Because of algorithmic similarity with MP2 calculation, the “resolution of the identity” approximation can also be used in CIS(D). In fact, RI-CIS(D) is orders of magnitudes more efficient than previously explained CIS(D) algorithms for effectively all molecules with more than a few atoms. Like in MP2, this is achieved by reducing the prefactor of the computational load. In fact, the overall cost still scales with the fifth power of the system size.
Presently in Q-Chem, RI approximation is supported for closed-shell restricted CIS(D) and open-shell unrestricted UCIS(D) energy calculations. The theta diagnostic is not implemented for RI-CIS(D).
6.6.3 SOS-CIS(D) Model
As in MP2 case, the accuracy of CIS(D) calculations can be improved by semi-empirically scaling the opposite-spin components of CIS(D) expression:
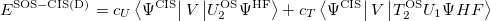 |
(6.24) |
with the corresponding ground state energy
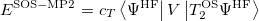 |
(6.25) |
More importantly, this SOS-CIS(D) energy can be evaluated with the 4th power of the molecular size by adopting Laplace transform technique [347]. Accordingly, SOS-CIS(D) can be applied to the calculations of excitation energies for relatively large molecules.
6.6.4 SOS-CIS(D ) Model
) Model
CIS(D) and its cousins explained in the above are all based on a second-order non-degenerate perturbative correction scheme on the CIS energy (“diagonalize-and-then-perturb” scheme). Therefore, they may fail when multiple excited states come close in terms of their energies. In this case, the system can be handled by applying quasi-degenerate perturbative correction scheme (“perturb-and-then-diagonalize” scheme). The working expression can be obtained by slightly modifying CIS(D) expression shown in Section 6.6.1 [349].
First, starting from Eq. (6.20), one can be explicitly write the CIS(D) energy as [350, 349]
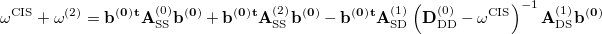 |
(6.26) |
To avoid the failures of the perturbation theory near degeneracies, the entire single and double blocks of the response matrix should be diagonalized. Because such a diagonalization is a non-trivial non-linear problem, an additional approximation from the binomial expansion of the 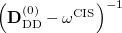 is further applied [349]:
is further applied [349]:
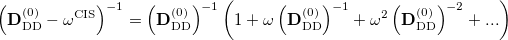 |
(6.27) |
The CIS(D) energy  is defined as the eigensolution of the response matrix with the zero-th order expansion of this equation. Namely,
is defined as the eigensolution of the response matrix with the zero-th order expansion of this equation. Namely,
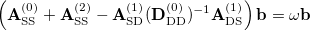 |
(6.28) |
Similar to SOS-CIS(D), SOS-CIS(D) theory is defined by taking the opposite-spin portions of this equation and then scaling them with two semi-empirical parameters [350]:
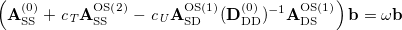 |
(6.29) |
Using the Laplace transform and the auxiliary basis expansion techniques, this can also be handled with a 4th-order scaling computational effort. In Q-Chem, an efficient 4th-order scaling analytical gradient of SOS-CIS(D) is also available. This can be used to perform excited state geometry optimizations on the electronically excited state surfaces.
6.6.5 CIS(D) Job Control and Examples
The legacy CIS(D) algorithm in Q-Chem is handled by the CCMAN/CCMAN2 modules of Q-Chem’s and shares many of the $rem options. RI-CIS(D), SOS-CIS(D), and SOS-CIS(D) do not depend on the coupled-cluster routines. Users who will not use this legacy CIS(D) method may skip to Section 6.6.6.
As with all post-HF calculations, it is important to ensure there are sufficient resources available for the necessary integral calculations and transformations. For CIS(D), these resources are controlled using the $rem variables CC_MEMORY, MEM_STATIC and MEM_TOTAL (see Section 5.7.7).
To request a CIS(D) calculation the METHOD $rem should be set to CIS(D) and the number of excited states to calculate should be specified by EE_STATES (or EE_SINGLETS and EE_TRIPLETS when appropriate). Alternatively, CIS(D) will be performed when EXCHANGE=HF, CORRELATION=CI and EOM_CORR=CIS(D). The SF-CIS(D) is invoked by using SF_STATES.
EE_STATES
Sets the number of excited state roots to find. For closed-shell reference, defaults into EE_SINGLETS. For open-shell references, specifies all low-lying states.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any excited states.
OPTIONS:
![$[i,j,k\ldots ]$](images/img-0690.png)
Find
excited states in the first irrep,
states in the second irrep etc.
RECOMMENDATION:
None
EE_SINGLETS
Sets the number of singlet excited state roots to find. Valid only for closed-shell references.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any excited states.
OPTIONS:
Find
RECOMMENDATION:
None
EE_TRIPLETS
Sets the number of triplet excited state roots to find. Valid only for closed-shell references.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any excited states.
OPTIONS:
Find
RECOMMENDATION:
None
SF_STATES
Sets the number of spin-flip target states roots to find.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any spin-flip states.
OPTIONS:
Find
RECOMMENDATION:
None
Note: It is a symmetry of a transition rather than that of a target state which is specified in excited state calculations. The symmetry of the target state is a product of the symmetry of the reference state and the transition. For closed-shell molecules, the former is fully symmetric and the symmetry of the target state is the same as that of transition, however, for open-shell references this is not so.
CC_STATE_TO_OPT
Specifies which state to optimize.
TYPE:
INTEGER ARRAY
DEFAULT:
None
OPTIONS:
[
optimize the
RECOMMENDATION:
None
Note: Since there are no analytic gradients for CIS(D), the symmetry should be turned off for geometry optimization and frequency calculations, and CC_STATE_TO_OPT should be specified assuming C symmetry, i.e., as [1,N] where N is the number of state to optimize (the states are numbered from 1).
symmetry, i.e., as [1,N] where N is the number of state to optimize (the states are numbered from 1).
Example 6.124 CIS(D) excitation energy calculation for ozone at the experimental ground state geometry C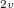
$molecule
0 1
O
O 1 RE
O 2 RE 1 A
RE=1.272
A=116.8
$end
$rem
JOBTYPE SP
METHOD CIS(D)
BASIS 6-31G*
N_FROZEN_CORE 3 use frozen core
EE_SINGLETS [2,2,2,2] find 2 lowest singlets in each irrep.
EE_TRIPLETS [2,2,2,2] find two lowest triplets in each irrep.
$end
Example 6.125 CIS(D) geometry optimization for the lowest triplet state of water. The symmetry is automatically turned off for finite difference calculations
$molecule
0 1
o
h 1 r
h 1 r 2 a
r 0.95
a 104.0
$end
$rem
jobtype opt
basis 3-21g
method cis(d)
ee_triplets 1 calculate one lowest triplet
cc_state_to_opt [1,1] optimize the lowest state (first state in first irrep)
$end
Example 6.126 CIS(D) excitation energy and transition property calculation (between all states) for ozone at the experimental ground state geometry C
$molecule
0 1
O
O 1 RE
O 2 RE 1 A
RE=1.272
A=116.8
$end
$rem
jobtype SP
BASIS 6-31G*
purcar 2 Non-spherical (6D)
method CIS(D)
ee_singlets [2,2,2,2]
ee_triplets [2,2,2,2]
cc_trans_prop 1
$end
6.6.6 RI-CIS(D), SOS-CIS(D), and SOS-CIS(D): Job Control
These methods are activated by setting the $rem keyword METHOD to RICIS(D), SOSCIS(D), and SOSCIS(D0), respectively. Other keywords are the same as in CIS method explained in Section 6.2.1. As these methods rely on the RI approximation, AUX_BASIS needs to be set by following the same guide as in RI-MP2 (Section 5.5).
METHOD
Excited state method of choice
TYPE:
STRING
DEFAULT:
None
OPTIONS:
RICIS(D)
Activate RI-CIS(D)
SOSCIS(D)
Activate SOS-CIS(D)
SOSCIS(D0)
Activate SOS-CIS(D
RECOMMENDATION:
None
CIS_N_ROOTS
Sets the number of excited state roots to find
TYPE:
INTEGER
DEFAULT:
0
Do not look for any excited states
OPTIONS:
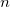
Looks for
RECOMMENDATION:
None
CIS_SINGLETS
Solve for singlet excited states (ignored for spin unrestricted systems)
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Solve for singlet states
FALSE
Do not solve for singlet states.
RECOMMENDATION:
None
CIS_TRIPLETS
Solve for triplet excited states (ignored for spin unrestricted systems)
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Solve for triplet states
FALSE
Do not solve for triplet states.
RECOMMENDATION:
None
SET_STATE_DERIV
Sets the excited state index for analytical gradient calculation for geometry optimizations and vibrational analysis with SOS-CIS(D
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Select the
RECOMMENDATION:
Check to see that the states do no change order during an optimization. For closed-shell systems, either CIS_SINGLETS or CIS_TRIPLETS must be set to false.
MEM_STATIC
Sets the memory for individual program modules
TYPE:
INTEGER
DEFAULT:
64
corresponding to 64 Mb
OPTIONS:
User-defined number of megabytes.
RECOMMENDATION:
At least
of MEM_STATIC is required (
: number of basis functions,
: size of a double precision storage, usually 8). Because a number of matrices with
size also need to be stored, 32–160 Mb of additional MEM_STATIC is needed.
MEM_TOTAL
Sets the total memory available to Q-Chem
TYPE:
INTEGER
DEFAULT:
2000
2 Gb
OPTIONS:
User-defined number of megabytes
RECOMMENDATION:
The minimum memory requirement of RI-CIS(D) is approximately MEM_STATIC + max
(
: number of excited states,
: number of auxiliary basis functions,
. SOS-CIS(D
more memory space is given. Like in RI-CIS(D), put as much memory as possible if you are not sure what to use. The actual memory size used in these calculations will be printed out in the output file to give a guide about the required memory.
AO2MO_DISK
Sets the scratch space size for individual program modules
TYPE:
INTEGER
DEFAULT:
2000
2 Gb
OPTIONS:
User-defined number of megabytes.
RECOMMENDATION:
The minimum disk requirement of RI-CIS(D) is approximately
. Again, the batching scheme will become more efficient with more available disk space. There is no simple formula for SOS-CIS(D) and SOS-CIS(D
SOS_FACTOR
Sets the scaling parameter
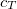
TYPE:
INTEGER
DEFAULT:
1300000
corresponding to 1.30
OPTIONS:
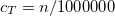
RECOMMENDATION:
Use the default
SOS_UFACTOR
Sets the scaling parameter
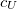
TYPE:
INTEGER
DEFAULT:
151
For SOS-CIS(D), corresponding to 1.51
140
For SOS-CIS(D
OPTIONS:
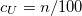
RECOMMENDATION:
Use the default
6.6.7 Examples
Example 6.127 Q-Chem input for an RI-CIS(D) calculation.
$molecule
0 1
C 0.667472 0.000000 0.000000
C -0.667472 0.000000 0.000000
H 1.237553 0.922911 0.000000
H 1.237553 -0.922911 0.000000
H -1.237553 0.922911 0.000000
H -1.237553 -0.922911 0.000000
$end
$rem
jobtype sp
method ricis(d)
basis aug-cc-pVDZ
mem_total 1000
mem_static 100
ao2mo_disk 1000
aux_basis rimp2-aug-cc-pVDZ
purecart 1111
cis_n_roots 10
cis_singlets true
cis_triplets false
$end
Example 6.128 Q-Chem input for an SOS-CIS(D) calculation.
$molecule
0 1
C -0.627782 0.141553 0.000000
O 0.730618 -0.073475 0.000000
H -1.133677 -0.033018 -0.942848
H -1.133677 -0.033018 0.942848
$end
$rem
jobtype sp
method soscis(d)
basis aug-cc-pVDZ
mem_total 1000
mem_static 100
ao2mo_disk 500000 ! 0.5 Terabyte of disk space available
aux_basis rimp2-aug-cc-pVDZ
purecart 1111
cis_n_roots 5
cis_singlets true
cis_triplets true
$end
Example 6.129 Q-Chem input for an SOS-CIS(D) geometry optimization on S surface.
surface.
$molecule
0 1
o
h 1 r
h 1 r 2 a
r 0.95
a 104.0
$end
$rem
jobtype = opt
method = soscis(d0)
basis = 6-31G**
aux_basis = rimp2-VDZ
purecart = 1112
set_state_deriv = 2
cis_n_roots = 5
cis_singlets = true
cis_triplets = false
$end