2.7 Running Q-Chem
Once installation is complete, and any necessary adjustments are made to the user account, the user is now able to run Q-Chem. There are several ways to invoke Q-Chem:
IQmol offers a fully integrated graphical interface for the Q-Chem package and includes a sophisticated input generator with contextual help which is able to guide you through the many Q-Chem options available. It also provides a molecular builder, job submission and monitoring tools, and is able to visualize molecular orbitals, densities and vibrational frequencies. For the latest version and download/installation instructions, please see the IQmol homepage (www.iqmol.org).
qchem command line shell script. The simple format for command line execution is given below. The remainder of this manual covers the creation of input files in detail.
Via a third-party GUI. The two most popular ones are:
A general web-based interface for electronic structure software, WebMO (see www.webmo.net).
Wavefunction’s Spartan user interface on some platforms. Contact Wavefunction (www.wavefun.com) or Q-Chem for full details of current availability.
Using the Q-Chem command line shell script (qchem) is straightforward provided Q-Chem has been correctly installed on your machine and the necessary environment variables have been set in your .cshrc, .profile, or equivalent login file. If done correctly, the necessary changes will have been made to the $PATH variable automatically on login so that Q-Chem can be invoked from your working directory.
The qchem shell script can be used in either of the following ways:
qchem infile outfile qchem infile outfile savename qchem --save infile outfile savename
where infile is the name of a suitably formatted Q-Chem input file (detailed in Chapter 3, and the remainder of this manual), and the outfile is the name of the file to which Q-Chem will place the job output information.
Note: If the outfile already exists in the working directory, it will be overwritten.
The use of the savename command line variable allows the saving of a few key scratch files between runs, and is necessary when instructing Q-Chem to read information from previous jobs. If the savename argument is not given, Q-Chem deletes all temporary scratch files at the end of a run. The saved files are in $QCSCRATCH/savename/, and include files with the current molecular geometry, the current molecular orbitals and density matrix and the current force constants (if available). The –save option in conjunction with savename means that all temporary files are saved, rather than just the few essential files described above. Normally this is not required. When $QCLOCALSCR has been specified, the temporary files will be stored there and copied to $QCSCRATCH/savename/ at the end of normal termination.
The name of the input parameters infile, outfile and save can be chosen at the discretion of the user (usual UNIX file and directory name restrictions apply). It maybe helpful to use the same job name for infile and outfile, but with varying suffixes. For example:
localhost-1> qchem water.in water.out &
invokes Q-Chem where the input is taken from water.in and the output is placed into water.out. The & places the job into the background so that you may continue to work in the current shell.
localhost-2> qchem water.com water.log water &
invokes Q-Chem where the input is assumed to reside in water.com, the output is placed into water.log and the key scratch files are saved in a directory $QCSCRATCH/water/.
Note: A checkpoint file can be requested by setting GUI=2 in the $rem section of the input. The checkpoint file name is determined by the GUIFILE environment variable which by default is set to ${input}.fchk
2.7.1 Running Q-Chem in parallel
The parallel execution of Q-Chem can be based on OpenMP threads on a single node, MPI processes over multiple cores or across multiple nodes, and a hybrid MPI+OpenMP scheme where each spawned MPI process creates OpenMP threads for computing parallel tasks. In the hybrid MPI+OpenMP scheme cross-node communication is handled by MPI protocol and intra-node communication is done implicitly using OpenMP threading for efficient utilization of shared memory SMP systems, which maps well with the current multi-core architecture in cluster computing.
As of the 4.2 release, the OpenMP parallelization is fully supported by HF/DFT, RIMP2, CC, EOM-CC, and ADC methods. The MPI parallel capability is available for SCF, DFT, CIS, and TDDFT methods. The hybrid MPI+OpenMP parallelization is introduced in 4.2 for HF/DFT energy and gradient calculations only. Table 2.1 summarizes the parallel capabilities of Q-Chem 4.2.
Method |
OpenMP |
MPI |
MPI+OpenMP |
HF energy & gradient |
yes |
yes |
yes |
DFT energy & gradient |
yes |
yes |
yes |
CDFT/CDFT-CI |
no |
no |
no |
RI-MP2 energy |
yes |
no |
no |
Attenuated RI-MP2 energy |
yes |
no |
no |
Integral transformation |
yes |
no |
no |
CCMAN & CCMAN2 methods |
yes |
no |
no |
ADC methods |
yes |
no |
no |
CIS energy & gradient |
no |
yes |
no |
TDDFT energy & gradient |
no |
yes |
no |
HF & DFT analytical Hessian |
no |
yes |
no |
To run Q-Chem calculation with OpenMP threads specify the number of threads ( nthreads ) using qchem command option -nt. Since each thread uses one CPU core, you should not specify more threads than the total number of available CPU cores for performance reason. When unspecified, the number of threads defaults to 1 (serial calculation).
qchem -nt nthreads infile outfile qchem -nt nthreads infile outfile save qchem -save -nt nthreads infile outfile save
Similarly, to run parallel calculations with MPI use the option -np to specify the number of MPI processes to be spawned.
qchem -np n infile outfile qchem -np n infile outfile savename qchem -save -np n infile outfile savename
where 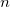 is the number of processors to use. If the -np switch is not given, Q-Chem will default to running locally on a single node.
is the number of processors to use. If the -np switch is not given, Q-Chem will default to running locally on a single node.
To run hybrid MPI+OpenMP HF/DFT calculations use combined options -np and -nt together, where -np followed by the number of MPI processes to be spawned and -nt followed by the number of OpenMP threads used in each MPI process.
qchem -np n -nt nthreads infile outfile qchem -np n -nt nthreads infile outfile savename qchem -save -np n -nt nthreads infile outfile savename
When the additional argument savename is specified, the temporary files for MPI-parallel Q-Chem are stored in $QCSCRATCH/savename.0 At the start of a job, any existing files will be copied into this directory, and on successful completion of the job, be copied to $QCSCRATCH/savename/ for future use. If the job terminates abnormally, the files will not be copied.
To run parallel Q-Chem using a batch scheduler such as PBS, users may need to set QCMPIRUN environment variable to point to the mpirun command used in the system. For further details users should read the $QC/README.Parallel file, and contact Q-Chem if any problems are encountered (support@q-chem.com).