10.7 Electrostatic Potentials
Q-Chem can evaluate electrostatic potentials on a grid of points. Electrostatic potential evaluation is controlled by the $rem variable IGDESP, as documented below.
IGDESP
Controls evaluation of the electrostatic potential on a grid of points. If enabled, the output is in an ASCII file, plot.esp, in the format
esp for each point.
TYPE:
INTEGER
DEFAULT:
none no electrostatic potential evaluation
OPTIONS:
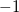
read grid input via the $plots section of the input deck
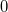
Generate the ESP values at all nuclear positions.
+
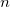
read
RECOMMENDATION:
None
The following example illustrates the evaluation of electrostatic potentials on a grid, note that IANLTY must also be set to 200.
Example 10.219 A job that evaluates the electrostatic potential for H on a 1 by 1 by 15 grid, along the bond axis. The output is in an ASCII file called plot.esp, which lists for each grid point,
on a 1 by 1 by 15 grid, along the bond axis. The output is in an ASCII file called plot.esp, which lists for each grid point,  ,
, 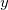 ,
,  , and the electrostatic potential.
, and the electrostatic potential.
$molecule
0 1
H 0.0 0.0 0.35
H 0.0 0.0 -0.35
$end
$rem
METHOD hf
BASIS 6-31g**
IANLTY 200
IGDESP -1
$end
$plots
plot the HOMO and the LUMO on a line
1 0.0 0.0
1 0.0 0.0
15 -3.0 3.0
0 0 0 0
0
$end
We can also compute the electrostatic potential for the transition density, which can be used, for example, to compute the Coulomb coupling in excitation energy transfer.
ESP_TRANS
Controls the calculation of the electrostatic potential of the transition density
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
compute the electrostatic potential of the excited state transition density
FALSE
compute the electrostatic potential of the excited state electronic density
RECOMMENDATION:
NONE
The electrostatic potential is a complicated object and it is not uncommon to model it using a simplified representation based on atomic charges. For this purpose it is well known that Mulliken charges perform very poorly. Several definitions of ESP-derived atomic charges have been given in the literature, however, most of them rely on a least-squares fitting of the ESP evaluated on a selection of grid points. Although these grid points are usually chosen so that the ESP is well modeled in the “chemically important” region, it still remains that the calculated charges will change if the molecule is rotated. Recently an efficient rotationally invariant algorithm was proposed [491] that sought to model the ESP not by direct fitting, but by fitting to the multipole moments. By doing so it was found that the fit to the ESP was superior to methods that relied on direct fitting of the ESP. The calculation requires the traceless form of the multipole moments and these are also printed out during the course of the calculations. To request these multipole-derived charges the following $rem option should be set:
MM_CHARGES
Requests the calculation of multipole-derived charges (MDCs).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculates the MDCs and also the traceless form of the multipole moments
RECOMMENDATION:
Set to TRUE if MDCs or the traceless form of the multipole moments are desired. The calculation does not take long.