1.3 Q-Chem Features
Quantum chemistry methods have proven invaluable for studying chemical and physical properties of molecules. The Q-Chem system brings together a variety of advanced computational methods and tools in an integrated ab initio software package, greatly improving the speed and accuracy of calculations being performed. In addition, Q-Chem will accommodate larger molecular structures than previously possible, with no loss in accuracy, thereby bringing the power of quantum chemistry to critical research projects for which this tool was previously unavailable. Below is a reverse-chronological listing of new features added to Q-Chem.
1.3.1 New Features in Q-Chem 5.1
Improved OpenMP parallelization for:
SCF vibrational frequency calculations (Z. Gan)
RIMP2 gradient (F. Rob, Joonho Lee, X. Feng, & E. Epifanovsky)
Complete active space self-consistent field (CASSCF) and adaptive sampling CI (D. Levine, M. Head-Gordon)
Tkatchenko-Scheffler van der Waals method (Section 5.7.4) and many-body dispersion method (Section 5.7.5) (D. Barton, Ka Un Lao, & R. DiStasio)
Enhancements to the coupled-cluster package:
Core/valence separation for EOM-CCSD core-level excited states (M. Vidal, A.I. Krylov, X. Feng, & S. Coriani), Section 7.7.5.
NTO analysis of two-photon transitions (K. Nanda & A.I. Krylov), Section 7.7.16.1.
NTO analysis of the complex-valued EOM wave functions (A.I. Krylov, W. Skomorowski), Section 7.7.16.
Analytic gradients for Cholesky-decomposed and resolution-of-identity CCSD and EOM-CCSD (X. Feng, A.I. Krylov).
Improved performance, reduced disk usage by coupled-cluster methods (E. Epifanovsky, I. Kaliman, & X. Feng).
New features in NTO analysis: Energies of NTOs (A.I. Krylov), Section 11.2.6.
Finite-difference evaluation of non-linear properties (M. de Wergifosse & A.I. Krylov), Section 11.14.2.
Poisson boundary conditions for SCF calculations (M. Coons & J. Herbert), Section 12.2.10.
Enables quantum chemistry calculations in an arbitrary (anisotropic and inhomogeneous) dielectric environment.
Nonequilibrium solvent corrections for vertical ionization energies.
Energy decomposition analysis (EDA):
EDA based on symmetry-adapted perturbation theory and constrained DFT (SAPT/cDFT-EDA), Section 13.13 (Ka Un Lao, K. Fenk, & J. Herbert)
ALMO-EDA for CIS and TDDFT/TDA excited states (Qinghui Ge, Yuezhi Mao, & M. Head-Gordon)
Perturbative ALMO-CTA and COVP analysis in EDA2 (Yuezhi Mao & M. Head-Gordon)
Analytic derivative couplings for computing excitation/vibration energy couplings within the ab initio Frenkel-Davydov exciton model (A. Morrison & J. Herbert), Section 13.15.3.
Hyperfine spin-spin couplings and nuclear electric quadrupole couplings, Section 11.13.3 (E. Berquist & D. Lambrecht)
Variational two-electron reduced-density-matrix (v2RDM) and v2RDM-driven complete active space self-consistent field (v2RDM-CASSCF) method (G. Gidofalvi, L. Koulias, J.W. Mullinax, & A.E. DePrince III)
Frozen and restrained potential energy scans, Section 10.4 (Yihan Shao)
Extended ESP charge fitting procedure to the computation of RESP charges (Yihan Shao)
1.3.2 New Features in Q-Chem 5.0
Enhancements to the coupled-cluster package:
Analytic gradients for Cholesky-decomposed CCSD and EOM-CCSD; efficiency improvement for canonical CCSD and EOM-CCSD gradients (X. Feng, E. Epifanovsky).
CAP-EOM-CCSD analytic gradients (Z. Benda and T.-C. Jagau) and Dyson orbitals for metastable states (T.-C. Jagau, A.I. Krylov), Section 7.7.6).
CAP-EOM-MP2 method (A. Kunitsa, K. Bravaya).
Evaluation of polarizabilities using CCSD and EOM-CCSD (EE and SF) wave functions using full derivative formulation (K. Nanda and A. Krylov, Section 7.7.16.4).
Evaluation of
 for EOM-CCSD wave functions (X. Feng).
for EOM-CCSD wave functions (X. Feng). Evaluation of NACs for EOM-CCSD wave functions (S. Faraji, A. Krylov, E. Epifanovski, X. Feng, Section 7.7.16.3).
Efficiency improvement and new multicore-parallel code for (T) correction (I. Kaliman).
New coupled-cluster based methods for core states (A. Krylov).
New capabilities for implicit solvation modeling:
PCM capabilities for computing vertical excitation, ionization, and electron attachment energies at EOM-CC and MP2 levels (Section 7.7.11).
State-specific equilibrium and non-equilibrium solvation for all orders and variants of ADC (J. M. Mewes and A. Dreuw; Section 7.8.7).
Poisson equation boundary conditions allowing use of an arbitrary, anisotropic dielectric function
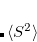 , with full treatment of volume polarization (M. P. Coons and J. M. Herbert; Section 12.2.10).
, with full treatment of volume polarization (M. P. Coons and J. M. Herbert; Section 12.2.10). Composite Model for Implicit Representation of Solvent (CMIRS), an accurate model for free energies of solvation (Section 12.2.6)
New density functionals (N. Mardirossian and M. Head-Gordon; Section 5.3):
GGA functionals: BEEF-vdW, HLE16, KT1, KT2, KT3, rVV10
Meta-GGA functionals: B97M-rV, BLOC, mBEEF, oTPSS, TM
Hybrids: CAM-QTP(00), CAM-QTP(01), HSE-HJS, LC-
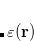 PBE08, MN15, rCAM-B3LYP, WC04, WP04
PBE08, MN15, rCAM-B3LYP, WC04, WP04 Double hybrids: B2GP-PLYP, DSD-PBEB95-D3, DSD-PBEP86-D3, DSD-PBEPBE-D3, LS1DH-PBE, PBE-QIDH, PTPSS-D3, PWPB95-D3
Grimme’s PBEh-3c “low-cost” composite method
rVV10 non-local correlation functional
Additional DFT developments:
New integral package for for computing effective core potential (ECP) integrals (S. C. McKenzie, E. Epifanovsky; Chapter 9).
More efficient analytic algorithms for energies and first derivatives.
Support for arbitrary projector angular momentum.
Support up to
 angular momentum in the basis set.
angular momentum in the basis set.
Analytic derivative couplings for the ab initio Frenkel-Davydov exciton model (A. F. Morrison and J. M. Herbert; Section 13.15.3).
New ALMO-based energy decomposition analysis (EDA) methods:
The second-generation ALMO-EDA methods for DFT (P. R. Horn, Y. Mao and M. Head-Gordon; Section 13.7)
The extension of ALMO-EDA to RIMP2 theory (J. Thirman and M. Head-Gordon; Section 13.8)
The “adiabatic" EDA method for decomposing changes in molecular properties (Y. Mao, P. R. Horn and M. Head-Gordon; Section 13.9)
Wave function correlation capabilities:
Coupled cluster valence bond (CCVB) method for describing open-shell molecules with strong spin correlations (D. W. Small and M. Head-Gordon; Section 6.15.2).
Implementation of coupled-cluster valence bond with singles and doubles (CCVB-SD) for closed-shell species (J. Lee, D. W. Small and M. Head-Gordon; Section 6.10.4).
Note: Several important changes in Q-Chem’s default settings have occurred since version 4.4. Core electrons are now frozen by default in most post-Hartree-Fock calculations; see Section 6.2. The keywords for calculation of SOCs and NACs were renamed for consistency between different methods. Some newer density functionals now use either the SG-2 or SG-3 quadrature grid by default, whereas all functionals used SG-1 by default in v. 4.4. Table 5.3 lists the default grid for various classes of functionals.
1.3.3 New Features in Q-Chem 4.4
occ-RI-K algorithm for the evaluation of exact exchange in energy and force calculations (S. Manzer, F. Rob and M. Head-Gordon; Section 4.6.9)
Combinatorially-optimized exchange-correlation functionals (N. Mardirossian and M. Head-Gordon; Section 5.3):
- B97M-V (range-separated hybrid, meta-GGA functional with VV10 non-local correlation)
B97M-V (meta-GGA functional with VV10 non-local correlation)
- B97X-V (range-separated hybrid functional with VV10 non-local correlation)
Implementation of new exchange-correlation functionals from the literature (N. Mardirossian and M. Head-Gordon; Section 5.3). These include:
MGGA_MS0, MGGA_MS1, MGGA_MS2, MGGA_MS2h, MGGA_MVS, MGGA_MVSh, PKZB, revTPSS, revTPSSh, SCAN, SCAN0, PBEsol, revPBE, revPBE0
N12, N12-SX, GAM, MN12-L, MN12-SX, MN15-L, dlDF
VV10, LC-VV10
B97-K, B97-D3(0), B97-3,
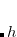 -HCTH, -HCTHh
-HCTH, -HCTHh SRC1-R1, SRC1-R2, SRC2-R1, SRC2-R2
B1LYP, B1PW91, MPW1K, LRC-BOP, BHH, BB1K, PW6B95, PWB6K, B2PLYP
Hessian-free minimum point verification (S. M. Sharada and M. Head-Gordon; Section 10.2.2)
Exciton-based excited-state models:
Ab initio Frenkel-Davydov model for coupled excitations in multi-chromophore systems (A. F. Morrison and J. M. Herbert; Section 13.15).
TDDFT for molecular interactions [TDDFT(MI)], a set of local excitation approximations for efficient TDDFT calculations in multi-chromophore systems and for single chromophores in the presence of explicit solvent molecules (J. Liu and J. M. Herbert; Section 13.16).
Improvements to many-body and XSAPT methods (K. U. Lao and J. M. Herbert)
Thermostats for ab initio molecular dynamics (R. P. Steele and J. M. Herbert).
Analytic energy gradient for the Ewald summation in QM/MM calculations (Z. C. Holden and J. M. Herbert)
Zeolite QM/MM methods (J. Gomes and M. Head-Gordon).
EOM-MP2 methods for excitation, ionization and electron attachment energies (A. Kunitsa and K. Bravaya; Section 7.7.9).
Evaluation of polarizabilities using CCSD and EOM-CCSD wave functions (Section 7.7.16.4, K. Nanda and A. I. Krylov)
Distributed-memory parallel implementation of CC and EOM-CC methods and performance improvements in disk-based algorithms (E. Epifanovsky, I. Kaliman, and A. I. Krylov)
Improvements to the maximum overlap method (MOM) for SCF calculations (A. T. B. Gilbert; Section 7.4).
Non-equilibrium PCM method to describe solvent effects in ADC excited-state calculations (J.-M. Mewes and A. Dreuw; Section 7.8.7).
Spin-flip ADC method (D. Lefrancois and A. Dreuw; Section 7.8.5).
1.3.4 New Features in Q-Chem 4.3
Analytic derivative couplings (i.e., non-adiabatic couplings) between electronic states computed at the CIS, spin-flip CIS, TDDFT, and spin-flip TDDFT levels (S. Fatehi, Q. Ou, J. E. Subotnik, X. Zhang, and J. M. Herbert; Section 10.6).
A third-generation (“+D3”) dispersion potential for XSAPT (K. U. Lao and J. M. Herbert; Section 13.12).
Non-equilibrium PCM for computing vertical excitation energies (at the TDDFT level) and ionization energies in solution (Z.-Q. You and J. M. Herbert; Section 12.2.2.3).
Spin-orbit couplings between electronic states for CC and EOM-CC wave functions (E. Epifanovsky, J. Gauss, and A. I. Krylov; Section 7.7.16.2).
PARI-K method for evaluation of exact exchange, which affords dramatic speed-ups for triple-
 and larger basis sets in hybrid DFT calculations (S. Manzer and M. Head-Gordon).
and larger basis sets in hybrid DFT calculations (S. Manzer and M. Head-Gordon). Transition moments and cross sections for two-photon absorption using EOM-CC wave functions (K. Nanda and A. I. Krylov; Section 7.7.16.1).
New excited-state analysis for ADC and CC/EOM-CC methods (M. Wormit; Section 11.2.6).
New Dyson orbital code for EOM-IP-CCSD and EOM-EA-CCSD (A. Gunina and A. I. Krylov; Section 7.7.23).
Transition moments, state dipole moments, and Dyson orbitals for CAP-EOM-CCSD (T.-C. Jagau and A. I. Krylov; Sections 7.7.6 and 7.7.23).
TAO-DFT: Thermally-assisted-occupation density functional theory (J.-D. Chai; Section 5.12).
MP2[V], a dual basis method that approximates the MP2 energy (J. Deng and A. Gilbert).
Iterative Hirshfeld population analysis for charged systems, and CM5 semi-empirical charge scheme (K. U. Lao and J. M. Herbert; Section 11.2.1).
New DFT functionals: (Section 5.3):
Long-range corrected functionals with empirical dispersion-:
M05-D, B97X-D3 and M06-D3 (Y.-S. Lin, K. Hui, and J.-D. Chai. PBE0_DH and PBE0_2 double-hybrid functionals (K. Hui and J.-D. Chai; Section 5.9).
AK13 (K. Hui and J.-D. Chai).
LFAs asymptotic correction scheme (P.-T. Fang and J.-D. Chai).
LDA/GGA fundamental gap using a frozen-orbital approximation (K. Hui and J.-D. Chai; Section 5.11).
1.3.5 New Features in Q-Chem 4.2
Input file changes:
New keyword METHOD simplifies input in most cases by replacing the pair of keywords EXCHANGE and CORRELATION (see Chapter 4).
Keywords for requesting excited-state calculations have been modified and simplified (see Chapter 7 for details).
Keywords for solvation models have been modified and simplified (see Section 12.2 for details).
New features for NMR calculations including spin-spin couplings (J. Kussmann, A. Luenser, and C. Ochsenfeld; Section 11.13.1).
New built-in basis sets (see Chapter 8).
New features and performance improvements in EOM-CC:
EOM-CC methods extended to treat meta-stable electronic states (resonances) via complex scaling and complex absorbing potentials (D. Zuev, T.-C. Jagau, Y. Shao, and A. I. Krylov; Section 7.7.6).
New features added to EOM-CC iterative solvers, such as methods for interior eigenvalues and user-specified guesses (D. Zuev; Section 7.7.12).
Multi-threaded parallel code for (EOM-)CC gradients and improved CCSD(T) performance.
New features and performance improvements in ADC methods (M. Wormit, A. Dreuw):
SM12 implicit solvation model (A. V. Marenich, D. G. Truhlar, and Y. Shao; Section 12.2.8.1).
Interface to NBO v. 6 (Section 11.3).
Optimization of MECPs between electronic states at the SOS-CIS(D) and TDDFT levels (X. Zhang and J. M. Herbert; Section 10.6.3).
ROKS method for
 SCF calculations of excited states (T. Kowalczyk and T. Van Voorhis; Section 7.5).
SCF calculations of excited states (T. Kowalczyk and T. Van Voorhis; Section 7.5). Fragment-based initial guess for SCF methods (Section 13.3).
Pseudo-fractional occupation number method for improved SCF convergence in small-gap systems (D. S. Lambrecht; Section 4.5.10).
Density embedding scheme (B. J. Albrecht, E. Berquist, and D. S. Lambrecht; Section 12.6).
New features and enhancements in fragment-based many-body expansion methods (K. U. Lao and J. M. Herbert):
Periodic boundary conditions with proper Ewald summation, for energies only (Z. C. Holden and J. M. Herbert; Section 12.3).
1.3.6 New Features in Q-Chem 4.1
Fundamental algorithms:
Improved parallel performance at all levels including new OpenMP capabilities for Hartree-Fock, DFT, MP2, and coupled cluster theory (Z. Gan, E. Epifanovsky, M. Goldey, and Y. Shao; Section 2.8).
Significantly enhanced ECP capabilities, including gradients and frequencies in all basis sets for which the energy can be evaluated (Y. Shao and M. Head-Gordon; Chap. 9).
SCF and DFT capabilities:
TDDFT energy with the M06, M08, and M11 series of functionals.
XYGJ-OS analytical energy gradient.
TDDFT/C-PCM excitation energies, gradient, and Hessian (J. Liu and W. Liang; Section 7.3.4).
Additional features in the maximum overlap method (MOM) approach for converging difficult SCF calculations (N. A. Besley; Section 4.5.6).
Wave function correlation capabilities:
RI and Cholesky decomposition implementation of all CC and EOM-CC methods enabling applications to larger systems with reduced disk and memory requirements and improved performance (E. Epifanovsky, X. Feng, D. Zuev, Y. Shao, and A. I. Krylov; Sections 6.8.5 and 6.8.6).
Attenuated MP2 theory in the aug-cc-pVDZ and aug-cc-pVTZ basis sets, which truncates two-electron integrals to cancel basis set superposition error, yielding results for intermolecular interactions that are much more accurate than standard MP2 in the same basis set (M. Goldey and M. Head-Gordon; Section 6.7).
Extended RAS-
 SF methodology for ground and excited states involving strong non-dynamical correlation (P. M. Zimmerman, D. Casanova, and M. Head-Gordon; Section 7.9).
SF methodology for ground and excited states involving strong non-dynamical correlation (P. M. Zimmerman, D. Casanova, and M. Head-Gordon; Section 7.9). Coupled cluster valence bond (CCVB) method for describing molecules with strong spin correlations (D. W. Small and M. Head-Gordon; Section 6.15.2).
Searching and scanning potential energy surfaces:
Potential energy surface scans (Y. Shao; Section 10.4).
Improvements in automatic transition structure searching via the “freezing string” method, including the ability to perform such calculations without a Hessian calculation (S. M. Sharada and M. Head-Gordon; Section 10.2.2).
Enhancements to partial Hessian vibrational analysis (N. A. Besley; Section 11.10.3).
Calculating and characterizing inter- and intramolecular interactions
Extension of EFP to macromolecules: fEFP approach (A. Laurent, D. Ghosh, A. I. Krylov, and L. V. Slipchenko; Section 12.5.3).
Symmetry-adapted perturbation theory level at the “SAPT0” level, for intermolecular interaction energy decomposition analysis into physically-meaningful components such as electrostatics, induction, dispersion, and exchange. An RI version is also available (L. D. Jacobson, J. M. Herbert; Section 13.11).
The “explicit polarization” (XPol) monomer-based SCF calculations to compute many-body polarization effects in linear-scaling time via charge embedding (Section 13.10), which can be combined either with empirical potentials (e.g., Lennard-Jones) for the non-polarization parts of the intermolecular interactions, or better yet, with SAPT for an ab initio approach called XSAPT that extends SAPT to systems containing more that two monomers (L. D. Jacobson and J. M. Herbert; Section 13.12).
Extension of the absolutely-localized molecular orbital (ALMO)-based energy decomposition analysis to unrestricted cases (P. R. Horn and M. Head-Gordon; Section 13.5).
Calculation of the populations of “effectively unpaired electrons” in low-spin state using DFT, a new method of evaluating localized atomic magnetic moments within Kohn-Sham without symmetry breaking, and Mayer-type bond order analysis with inclusion of static correlation effects (E. I. Proynov; Section 11.17).
Quantum transport calculations including electron transmission functions and electron tunneling currents under applied bias voltage (B. D. Dunietz and N. Sergueev; Section 11.18).
Searchable online version of the Q-Chem PDF manual (J. M. Herbert and E. Epifanovsky).
1.3.7 New Features in Q-Chem 4.0.1
Remote submission capability in IQmol (A. T. B. Gilbert).
Scaled nuclear charge and charge-cage stabilization capabilities (T. Kús and A. I. Krylov; Section 7.7.7).
Calculations of excited state properties including transition dipole moments between different excited states in CIS and TDDFT as well as couplings for electron and energy transfer (Z.-Q. You and C.-P. Hsu; Section 11.16).
1.3.8 New Features in Q-Chem 4.0
New exchange-correlation functionals (Section 5.3):
Density-functional dispersion using Becke and Johnson’s XDM model in an efficient, analytic form (Z. Gan, E. I. Proynov, and J. Kong; Section 5.7.3).
Van der Waals density functionals vdW-DF-04 and vdW-DF-10 of Langreth and coworkers (O. Vydrov; Section 5.7.1).
VV09 and VV10, new analytic dispersion functionals (O. Vydrov, T. Van Voorhis; Section 5.7.1)
DFT-D3 empirical dispersion methods for non-covalent interactions (S.-P. Mao and J.-D. Chai; Section 5.7.2).
- B97X-2, a double-hybrid functional based on the long-range corrected B97 functional, with improved accounting for medium- and long-range interactions (J.-D. Chai and M. Head-Gordon; Section 5.9).
XYGJ-OS, a double-hybrid functional for predictions of non-bonded interactions and thermochemistry at nearly chemical accuracy (X. Xu, W. A. Goddard, and Y. Jung; Section 5.9).
Short-range corrected functional for calculation of near-edge X-ray absorption spectra (N. A. Besley; Section 7.10.1).
LB94 asymptotically-corrected exchange-correlation functional for TDDFT (Y.-C. Su and J.-D. Chai; Section 5.10.1).
Non-dynamical correlation in DFT with an efficient RI implementation of the Becke05 model in a fully analytic formulation (E. I. Proynov, Y. Shao, F. Liu, and J. Kong; Section 5.3).
TPSS and its hybrid version TPSSh, and rPW86 (F. Liu and O. Vydrov).
Double-hybrid functional B2PLYP-D (J.-D. Chai).
Hyper-GGA functional MCY2 from Mori-Sánchez, Cohen, and Yang (F. Liu).
SOGGA, SOGGA11 and SOGGA11-X family of GGA functionals (R. Peverati, Y. Zhao, and D. G. Truhlar).
M08-HX and M08-SO suites of high HF exchange meta-GGA functionals (Y. Zhao and D. G. Truhlar).
M11-L and M11 suites of meta-GGA functionals (R. Peverati, Y. Zhao, D. G. Truhlar).
Improved DFT algorithms:
Multi-resolution exchange-correlation (mrXC) for fast calculation of grid-based XC quadrature (S. T. Brown, C.-M. Chang, and J. Kong; Section 5.5.4).
Efficient computation of the XC part of the dual basis DFT (Z. Gan and J. Kong; Section 4.4.5).
Fast DFT calculation with “triple jumps” between different sizes of basis set and grid, and different levels of functional (J. Deng, A. T. B. Gilbert, and P. M. W. Gill; Section 4.8).
Faster DFT and HF calculation with an atomic resolution-of-identity algorithm (A. Sodt and M. Head-Gordon; Section 4.6.6).
Post-Hartree–Fock methods:
Significantly enhanced coupled-cluster code rewritten for better performance on multi-core architectures, including energy and gradient calculations with CCSD and energy calculations with EOM-EE/SF/IP/EA-CCSD, and CCSD(T) energy calculations (E. Epifanovsky, M. Wormit, T. Kús, A. Landau, D. Zuev, K. Khistyaev, I. Kaliman, A. I. Krylov, and A. Dreuw; Chaps. 6 and 7).
Fast and accurate coupled-cluster calculations with frozen natural orbitals (A. Landau, D. Zuev, and A. I. Krylov; Section 6.11).
Correlated excited states with the perturbation-theory based, size-consistent ADC scheme (M. Wormit and A. Dreuw; Section 7.8).
Restricted active space, spin-flip method for multi-configurational ground states and multi-electron excited states (P. M. Zimmerman, F. Bell, D. Casanova, and M. Head-Gordon; Section 7.2.4).
Post-Hartree–Fock methods for describing strong correlation:
TDDFT for excited states:
Nuclear gradients for TDDFT (Z. Gan, C.-P. Hsu, A. Dreuw, M. Head-Gordon, and J. Kong; Section 7.3.1).
Direct coupling of charged states for study of charge transfer reactions (Z.-Q. You and C.-P. Hsu; Section 11.16.2).
Analytical excited-state Hessian for TDDFT within the Tamm-Dancoff approximation (J. Liu and W. Liang; Section 7.3.5).
Self-consistent excited-states with the maximum overlap method (A. T. B. Gilbert, N. A. Besley, and P. M. W. Gill; Section 7.4).
Calculation of reactions via configuration interactions of charge-constrained states computed with constrained DFT (Q. Wu, B. Kaduk and T. Van Voorhis; Section 5.13).
Overlap analysis of the charge transfer in a TDDFT excited state (N. A. Besley; Section 7.3.2).
Localizing diabatic states with Boys or Edmiston-Ruedenberg localization, for charge or energy transfer (J. E Subotnik, R. P. Steele, N. Shenvi, and A. Sodt; Section 11.16.1.2).
Non-collinear formalism for spin-flip TDDFT (Y. Shao, Y. A. Bernard, and A. I. Krylov; Section 7.3)
Solvation and condensed-phase modeling
Smooth free energy surface for solvated molecules via SWIG-PCMs, for QM and QM/MM calculations, including a linear-scaling QM/MM/PCM algorithm (A. W. Lange and J. M. Herbert; Sections 12.2.2 and 12.2.4).
Klamt’s COSMO solvation model with DFT energy and gradient (Y. Shao; Section 12.2.7).
Polarizable explicit solvent via EFP, for ground- and excited-state calculations at the DFT/TDDFT and CCSD/EOM-CCSD levels, as well as CIS and CIS(D). A library of effective fragments for common solvents is also available, along with energy and gradient for EFP–EFP calculations (V. Vanovschi, D. Ghosh, I. Kaliman, D. Kosenkov, C. F. Williams, J. M. Herbert, M. S. Gordon, M. W. Schmidt, Y. Shao, L. V. Slipchenko, and A. I. Krylov; Section 12.5).
Optimizations, vibrations, and dynamics:
“Freezing” and “growing” string methods for efficient automated reaction-path finding (A. Behn, P. M. Zimmerman, A. T. Bell, and M. Head-Gordon; Section 10.2.1).
Improved robustness of the intrinsic reaction coordinate (IRC)-following code (M. Head-Gordon).
Quantum-mechanical treatment of nuclear motion at equilibrium via path integrals (R. P. Steele; Section 10.8).
Calculation of local vibrational modes of interest with partial Hessian vibrational analysis (N. A. Besley; Section 11.10.3).
Accelerated ab initio molecular dynamics MP2 and/or dual-basis methods, based on
 -vector extrapolation (R. P. Steele; Section 4.7.2).
-vector extrapolation (R. P. Steele; Section 4.7.2). Quasi-classical ab initio molecular dynamics (D. S. Lambrecht and M. Head-Gordon; Section 10.7.5).
Fragment-based methods:
Symmetry-adapted perturbation theory (SAPT) for computing and analyzing dimer interaction energies (L. D. Jacobson, M. A. Rohrdanz, and J. M. Herbert; Section 13.11).
Many-body generalization of SAPT (“XSAPT”), with empirical dispersion corrections for high accuracy and low cost in large clusters (L. D. Jacobson, K. U. Lao, and J. M. Herbert; Section 13.12).
Methods based on a truncated many-body expansion, including the fragment molecular orbital (FMO) method (K. U. Lao and J. M. Herbert; Section 13.14).
Properties and wave function analysis:
Analysis of metal oxidation states via localized orbital bonding analysis (A. J. W. Thom, E. J. Sundstrom, and M. Head-Gordon; Section 11.2.4).
Hirshfeld population analysis (S. Yeganeh; Section 11.2.1).
Visualization of non-covalent bonding using Johnson and Yang’s NCI algorithm (Y. Shao; Section 11.5.5).
Electrostatic potential on a grid for transition densities (Y. Shao; Section 11.5.6).
Support for modern computing platforms
Efficient multi-threaded parallel performance for CC, EOM, and ADC methods.
Better performance for multi-core systems with shared-memory parallel DFT and Hartree-Fock (Z. Gan, Y. Shao, and J. Kong) and RI-MP2 (M. Goldey and M. Head-Gordon; Section 6.14).
Accelerated RI-MP2 calculation on GPUs (R. Olivares-Amaya, M. Watson, R. Edgar, L. Vogt, Y. Shao, and A. Aspuru-Guzik; Section 6.6.4).
Graphical user interfaces:
Input file generation, Q-Chem job submission, and visualization is supported by IQmol, a fully integrated GUI developed by Andrew Gilbert. IQmol is a free software and does not require purchasing a Q-Chem license. See www.iqmol.org for details and installation instructions.
Other graphical interfaces are also available, including MolDen, MacMolPlt, and Avogadro (Chapter 11 and elsewhere).
1.3.9 Summary of Features in Q-Chem versions 3. x
x
DFT functionals and algorithms:
Long-ranged corrected (LRC) functionals, also known as range-separated hybrid functionals (M. A. Rohrdanz and J. M. Herbert)
Constrained DFT (Q. Wu and T. Van Voorhis)
Grimme’s “DFT-D” empirical dispersion corrections (C.-D. Sherrill)
“Incremental” DFT method that significantly accelerates exchange-correlation quadrature in later SCF cycles (S. T. Brown)
Efficient SG-0 quadrature grid with approximately half the number of grid points relative to SG-1 (S.-H. Chien)
Solvation models:
SM8 model (A. V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer, and D. G. Truhlar)
Onsager reaction-field model (C.-L. Cheng, T. Van Voorhis, K. Thanthiriwatte, and S. R. Gwaltney)
Chipman’s SS(V)PE model (S. T. Brown)
Second-order perturbation theory algorithms for ground and excited states:
Dual-basis RIMP2 energy and analytical gradient (R. P. Steele, R. A. DiStasio Jr., and M. Head-Gordon)
O2 energy and gradient (R. C. Lochan and M. Head-Gordon)
SOS-CIS(D), SOS-CIS(D
 ), and RI-CIS(D) for excited states (D. Casanova, Y. M. Rhee, and M. Head-Gordon)
), and RI-CIS(D) for excited states (D. Casanova, Y. M. Rhee, and M. Head-Gordon) Efficient resolution-of-identity (RI) implementations of MP2 and SOS-MP2 (including both energies and gradients), and of RI-TRIM and RI-CIS(D) energies (Y. Jung, R. A. DiStasio, Jr., R. C. Lochan, and Y. M. Rhee)
Coupled-cluster methods (P. A. Pieniazek, E. Epifanovsky, A. I. Krylov):
IP-CISD and EOM-IP-CCSD energy and gradient
Multi-threaded (OpenMP) parallel coupled-cluster calculations
Potential energy surface crossing minimization with CCSD and EOM-CCSD methods (E. Epifanovsky)
Dyson orbitals for ionization from the ground and excited states within CCSD and EOM-CCSD methods (M. Oana)
QM/MM methods (H. L. Woodcock, A. Ghysels, Y. Shao, J. Kong, and H. B. Brooks)
Q-Chem/Charmm interface (H. L. Woodcock)
Full QM/MM Hessian evaluation and approximate mobile-block-Hessian evaluation
Two-layer ONIOM model (Y. Shao).
Integration with the Molaris simulation package (E. Rosta).
Improved two-electron integrals package
Rewrite of the Head-Gordon–Pople algorithm for modern computer architectures (Y. Shao)
Fourier Transform Coulomb method for linear-scaling construction of the Coulomb matrix, even for basis sets with high angular moment and diffuse functions (L. Fusti-Molnar)
Dual basis self-consistent field calculations, offering an order-of-magnitude reduction in the cost of large-basis DFT calculations (J. Kong and R. P. Steele)
Enhancements to the correlation package including:
Most extensive range of EOM-CCSD methods available including EOM-SF-CCSD, EOM-EE-CCSD, EOM-DIP-CCSD, EOM-IP/EA-CCSD (A. I. Krylov).
Available for RHF, UHF, and ROHF references.
Analytic gradients and properties calculations (permanent and transition dipoles etc..).
Full use of Abelian point-group symmetry.
Coupled-cluster perfect-paring methods applicable to systems with
 active electrons (M. Head-Gordon)
active electrons (M. Head-Gordon) Transition structure search using the “growing string” algorithm (A. Heyden and B. Peters):
Ab initio molecular dynamics (J. M. Herbert)
Linear scaling properties for large systems (J. Kussmann, C. Ochsenfeld):
NMR chemical shifts
Static and dynamic polarizabilities
Static hyper-polarizabilities, optical rectification, and electro-optical Pockels effect
Anharmonic frequencies (C. Y. Lin)
Wave function analysis tools:
Analysis of intermolecular interactions with ALMO-EDA (R. Z. Khaliullin and M. Head-Gordon)
Electron transfer analysis (Z.-Q. You and C.-P. Hsu)
Spin densities at the nuclei (V. A. Rassolov)
Position, momentum, and Wigner intracules (N. A. Besley and D. P. O’Neill)
Graphical user interface options:
IQmol, a fully integrated GUI. IQmol includes input file generator and contextual help, molecular builder, job submission tool, and visualization kit (molecular orbital and density viewer, frequencies, etc). For the latest version and download/installation instructions, please see the IQmol homepage (www.iqmol.org).
Seamless integration with the Spartan package (see www.wavefun.com).
Support for several other public-domain visualization programs:
WebMO
www.webmo.netAvogadro
https://avogadro.ccMolDen
http://www.cmbi.ru.nl/moldenMacMolPlt (via a MolDen-formatted input file)
https://brettbode.github.io/wxmacmolpltJMol
www.sourceforge.net/project/showfiles.php?group_id= 23629release_id=66897
1.3.10 Summary of Features Prior to Q-Chem 3.0
Efficient algorithms for large-molecule density functional calculations:
CFMM for linear scaling Coulomb interactions (energies and gradients) (C. A. White).
Second-generation J-engine and J-force engine (Y. Shao).
LinK for exchange energies and forces (C. Ochsenfeld and C. A. White).
Linear scaling DFT exchange-correlation quadrature.
Local, gradient-corrected, and hybrid DFT functionals:
Slater, Becke, GGA91 and Gill ‘96 exchange functionals.
VWN, PZ81, Wigner, Perdew86, LYP and GGA91 correlation functionals.
EDF1 exchange-correlation functional (R. Adamson).
B3LYP, B3P and user-definable hybrid functionals.
Analytical gradients and analytical frequencies.
SG-0 standard quadrature grid (S.-H. Chien).
Lebedev grids up to 5294 points (S. T. Brown).
High level wave function-based electron correlation methods
Efficient semi-direct MP2 energies and gradients.
MP3, MP4, QCISD, CCSD energies.
OD and QCCD energies and analytical gradients.
Triples corrections (QCISD(T), CCSD(T) and OD(T) energies).
CCSD(2) and OD(2) energies.
Active space coupled cluster methods: VOD, VQCCD, VOD(2).
Local second order Møller-Plesset (MP2) methods (DIM and TRIM).
Improved definitions of core electrons for post-HF correlation (V. A. Rassolov).
Extensive excited state capabilities:
CIS energies, analytical gradients and analytical frequencies.
CIS(D) energies.
Time-dependent density functional theory energies (TDDFT).
Coupled cluster excited state energies, OD and VOD (A. I. Krylov).
Coupled-cluster excited-state geometry optimizations.
Coupled-cluster property calculations (dipoles, transition dipoles).
Spin-flip calculations for CCSD and TDDFT excited states (A. I. Krylov and Y. Shao).
High performance geometry and transition structure optimization (J. Baker):
Optimizes in Cartesian, Z-matrix or delocalized internal coordinates.
Impose bond angle, dihedral angle (torsion) or out-of-plane bend constraints.
Freezes atoms in Cartesian coordinates.
Constraints do not need to be satisfied in the starting structure.
Geometry optimization in the presence of fixed point charges.
Intrinsic reaction coordinate (IRC) following code.
Evaluation and visualization of molecular properties
Onsager, SS(V)PE and Langevin dipoles solvation models.
Evaluate densities, electrostatic potentials, orbitals over cubes for plotting.
Natural Bond Orbital (NBO) analysis.
Attachment/detachment densities for excited states via CIS, TDDFT.
Vibrational analysis after evaluation of the nuclear coordinate Hessian.
Isotopic substitution for frequency calculations (R. Doerksen).
NMR chemical shifts (J. Kussmann).
Atoms in Molecules (AIMPAC) support (J. Ritchie).
Stability analysis of SCF wave functions (Y. Shao).
Calculation of position and momentum molecular intracules A. Lee, N. A. Besley, and D. P. O’Neill).
Flexible basis set and effective core potential (ECP) functionality: (Ross Adamson and Peter Gill)
Wide range of built-in basis sets and ECPs.
Basis set superposition error correction.
Support for mixed and user-defined basis sets.
Effective core potentials for energies and gradients.
Highly efficient PRISM-based algorithms to evaluate ECP matrix elements.
Faster and more accurate ECP second derivatives for frequencies.