7.3 Time-Dependent Density Functional Theory (TDDFT)
7.3.1 Brief Introduction to TDDFT
Excited states may be obtained from density functional theory by time-dependent density functional theory,[Casida(1995), Dreuw and Head-Gordon(2005)] which calculates poles in the response of the ground state density to a time-varying applied electric field. These poles are Bohr frequencies, or in other words the excitation energies. Operationally, this involves solution of an eigenvalue equation
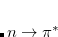 |
(7.15) |
where the elements of the matrix 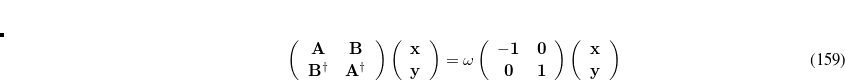 similar to those used at the CIS level, Eq. eq:orbital-Hessian, but with an exchange-correlation correction.[Hirata and Head-Gordon(1999a)] Elements of
similar to those used at the CIS level, Eq. eq:orbital-Hessian, but with an exchange-correlation correction.[Hirata and Head-Gordon(1999a)] Elements of 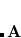 are similar. Equation eq:TDDFT is solved iteratively for the lowest few excitation energies,
are similar. Equation eq:TDDFT is solved iteratively for the lowest few excitation energies, 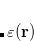 . Alternatively, one can make a CIS-like Tamm-Dancoff approximation (TDA),[Hirata and Head-Gordon(1999b)] in which the “de-excitation” amplitudes
. Alternatively, one can make a CIS-like Tamm-Dancoff approximation (TDA),[Hirata and Head-Gordon(1999b)] in which the “de-excitation” amplitudes 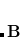 are neglected, the matrix is not required, and Eq. eq:TDDFT reduces to
are neglected, the matrix is not required, and Eq. eq:TDDFT reduces to 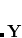 .
.
TDDFT is popular because its computational cost is roughly similar to that of the simple CIS method, but a description of differential electron correlation effects is implicit in the method. It is advisable to only employ TDDFT for low-lying valence excited states that are below the first ionization potential of the molecule,[Casida(1995)] or more conservatively, below the first Rydberg state, and in such cases the valence excitation energies are often remarkably improved relative to CIS, with an accuracy of 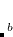 0.3 eV for many functionals.[Laurent and Jacquemin(2013)] The calculation of the nuclear gradients of full TDDFT and within the TDA is implemented.[Liu et al.(2010)Liu, Gan, Shao, Hsu, Dreuw, Head-Gordon, Miller, Brooks, Yu, Furlani, and Kong]
0.3 eV for many functionals.[Laurent and Jacquemin(2013)] The calculation of the nuclear gradients of full TDDFT and within the TDA is implemented.[Liu et al.(2010)Liu, Gan, Shao, Hsu, Dreuw, Head-Gordon, Miller, Brooks, Yu, Furlani, and Kong]
On the other hand, standard density functionals do not yield a potential with the correct long-range Coulomb tail, owing to the so-called self-interaction problem, and therefore excitation energies corresponding to states that sample this tail (e.g., diffuse Rydberg states and some charge transfer excited states) are not given accurately.[Casida et al.(1998)Casida, Jamorski, Casida, and Salahub, Tozer and Handy(1998), Lange and Herbert(2007)] The extent to which a particular excited state is characterized by charge transfer can be assessed using an a spatial overlap metric proposed by Peach, Benfield, Helgaker, and Tozer (PBHT).[Peach et al.(2008)Peach, Benfield, Helgaker, and Tozer] (However, see Ref. Richard:2011 for a cautionary note regarding this metric.)
Standard TDDFT also does not yield a good description of static correlation effects (see Section 6.10), because it is based on a single reference configuration of Kohn-Sham orbitals. Recently, a new variation of TDDFT called spin-flip (SF) DFT was developed by Yihan Shao, Martin Head-Gordon and Anna Krylov to address this issue.[Shao et al.(2003)Shao, Head-Gordon, and Krylov] SF-DFT is different from standard TDDFT in two ways:
The reference is a high-spin triplet (quartet) for a system with an even (odd) number of electrons;
One electron is spin-flipped from an alpha Kohn-Sham orbital to a beta orbital during the excitation.
SF-DFT can describe the ground state as well as a few low-lying excited states, and has been applied to bond-breaking processes, and di- and tri-radicals with degenerate or near-degenerate frontier orbitals. Recently, we also implemented[Bernard et al.(2012a)Bernard, Shao, and Krylov] a SF-DFT method with a non-collinear exchange-correlation potential from Tom Ziegler et al.,[Wang and Ziegler(2004), Seth et al.(2011)Seth, Mazur, and Ziegler] which is in many case an improvement over collinear SF-DFT.[Shao et al.(2003)Shao, Head-Gordon, and Krylov] Recommended functionals for SF-DFT calculations are 5050 and PBE50 (see Ref. Shao:2012 for extensive benchmarks). See also Section 7.7.3 for details on wave function-based spin-flip models.
7.3.2 TDDFT within a Reduced Single-Excitation Space
Much of chemistry and biology occurs in solution or on surfaces. The molecular environment can have a large effect on electronic structure and may change chemical behavior. Q-Chem is able to compute excited states within a local region of a system through performing the TDDFT (or CIS) calculation with a reduced single excitation subspace,[Besley(2004)] in which some of the amplitudes 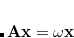 in Eq. eq:TDDFT are excluded. (This is implemented within the TDA, so
in Eq. eq:TDDFT are excluded. (This is implemented within the TDA, so  .) This allows the excited states of a solute molecule to be studied with a large number of solvent molecules reducing the rapid rise in computational cost. The success of this approach relies on there being only weak mixing between the electronic excitations of interest and those omitted from the single excitation space. For systems in which there are strong hydrogen bonds between solute and solvent, it is advisable to include excitations associated with the neighboring solvent molecule(s) within the reduced excitation space.
.) This allows the excited states of a solute molecule to be studied with a large number of solvent molecules reducing the rapid rise in computational cost. The success of this approach relies on there being only weak mixing between the electronic excitations of interest and those omitted from the single excitation space. For systems in which there are strong hydrogen bonds between solute and solvent, it is advisable to include excitations associated with the neighboring solvent molecule(s) within the reduced excitation space.
The reduced single excitation space is constructed from excitations between a subset of occupied and virtual orbitals. These can be selected from an analysis based on Mulliken populations and molecular orbital coefficients. For this approach the atoms that constitute the solvent needs to be defined. Alternatively, the orbitals can be defined directly. The atoms or orbitals are specified within a $solute block. These approach is implemented within the TDA and has been used to study the excited states of formamide in solution,[Besley et al.(2004)Besley, Oakley, Cowan, and Hirst] CO on the Pt(111) surface,[Besley(2005)] and the tryptophan chromophore within proteins.[Rogers et al.(2005)Rogers, Besley, O’Shea, and Hirst]
7.3.3 Job Control for TDDFT
Input for time-dependent density functional theory calculations follows very closely the input already described for the uncorrelated excited state methods described in the previous section (in particular, see Section 7.2.8). There are several points to be aware of:
The exchange and correlation functionals are specified exactly as for a ground state DFT calculation, through EXCHANGE and CORRELATION.
If RPA is set to TRUE, a “full” TDDFT calculation will be performed, however the default value is RPA = FALSE, which invokes the TDA,[Hirata and Head-Gordon(1999b)] in which the de-excitation amplitudes
in Eq. eq:TDDFT are neglected, which is usually a good approximation for excitation energies, although oscillator strengths within the TDA no longer formally satisfy the Thomas-Reiche-Kuhn sum rule.[Casida(1995)] For RPA = TRUE, a TDA calculation is performed first and used as the initial guess for the full TDDFT calculation. The TDA calculation can be skipped altogether using RPA = 2 If SPIN_FLIP is set to TRUE when performing a TDDFT calculation, a SF-DFT calculation will also be performed. At present, SF-DFT is only implemented within TDDFT/TDA so RPA must be set to FALSE. Remember to set the spin multiplicity to 3 for systems with an even-number of electrons (e.g., diradicals), and 4 for odd-number electron systems (e.g., triradicals).
TRNSS
Controls whether reduced single excitation space is used.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use full excitation space.
OPTIONS:
TRUE
Use reduced excitation space.
RECOMMENDATION:
None
TRTYPE
Controls how reduced subspace is specified.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Select orbitals localized on a set of atoms.
2
Specify a set of orbitals.
3
Specify a set of occupied orbitals, include excitations to all virtual orbitals.
RECOMMENDATION:
None
N_SOL
Specifies number of atoms or orbitals in the $solute section.
TYPE:
INTEGER
DEFAULT:
No default.
OPTIONS:
User defined.
RECOMMENDATION:
None
CISTR_PRINT
Controls level of output.
TYPE:
LOGICAL
DEFAULT:
FALSE
Minimal output.
OPTIONS:
TRUE
Increase output level.
RECOMMENDATION:
None
CUTOCC
Specifies occupied orbital cutoff.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
0-200
CUTOFF = CUTOCC/100
RECOMMENDATION:
None
CUTVIR
Specifies virtual orbital cutoff.
TYPE:
INTEGER
DEFAULT:
0
No truncation
OPTIONS:
0-100
CUTOFF = CUTVIR/100
RECOMMENDATION:
None
PBHT_ANALYSIS
Controls whether overlap analysis of electronic excitations is performed.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform overlap analysis.
TRUE
Perform overlap analysis.
RECOMMENDATION:
None
PBHT_FINE
Increases accuracy of overlap analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
TRUE
Increase accuracy of overlap analysis.
RECOMMENDATION:
None
SRC_DFT
Selects form of the short-range corrected functional.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
1
SRC1 functional.
2
SRC2 functional.
RECOMMENDATION:
None
OMEGA
Sets the Coulomb attenuation parameter for the short-range component.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:

Corresponding to
, in units of bohr
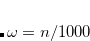
RECOMMENDATION:
None
OMEGA2
Sets the Coulomb attenuation parameter for the long-range component.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to
, in units of bohr
RECOMMENDATION:
None
HF_SR
Sets the fraction of Hartree-Fock exchange at
.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to HF_SR =
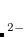
RECOMMENDATION:
None
HF_LR
Sets the fraction of Hartree-Fock exchange at
.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to HF_LR =
RECOMMENDATION:
None
WANG_ZIEGLER_KERNEL
Controls whether to use the Wang-Ziegler non-collinear exchange-correlation kernel in a SF-DFT calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not use non-collinear kernel.
TRUE
Use non-collinear kernel.
RECOMMENDATION:
None
7.3.4 TDDFT Coupled with C-PCM for Excitation Energies and Properties Calculations
As described in Section 12.2 (and especially Section 12.2.2), continuum solvent models such as C-PCM allow one to include solvent effect in the calculations. TDDFT/C-PCM allows excited-state modeling in solution. Q-Chem also features TDDFT coupled with C-PCM which extends TDDFT to calculations of properties of electronically-excited molecules in solution. In particular, TDDFT/C-PCM allows one to perform geometry optimization and vibrational analysis.[Liu and Liang(2013)]
When TDDFT/C-PCM is applied to calculate vertical excitation energies, the solvent around vertically excited solute is out of equilibrium. While the solvent electron density equilibrates fast to the density of the solute (electronic response), the relaxation of nuclear degrees of freedom (e.g., orientational polarization) takes place on a slower timescale. To describe this situation, an optical dielectric constant is employed. To distinguish between equilibrium and non-equilibrium calculations, two dielectric constants are used in these calculations: a static constant (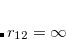 ), equal to the equilibrium bulk value, and a fast constant (
), equal to the equilibrium bulk value, and a fast constant ( ) related to the response of the medium to high frequency perturbations. For vertical excitation energy calculations (corresponding to the unrelaxed solvent nuclear degrees of freedom), it is recommended to use the optical dielectric constant for ), whereas for the geometry optimization and vibrational frequency calculations, the static dielectric constant should be used.[Liu and Liang(2013)]
) related to the response of the medium to high frequency perturbations. For vertical excitation energy calculations (corresponding to the unrelaxed solvent nuclear degrees of freedom), it is recommended to use the optical dielectric constant for ), whereas for the geometry optimization and vibrational frequency calculations, the static dielectric constant should be used.[Liu and Liang(2013)]
The example below illustrates TDDFT/C-PCM calculations of vertical excitation energies. More information concerning the C-PCM and the various PCM job control options can be found in Section 12.2.
Example 7.105 TDDFT/C-PCM low-lying vertical excitation energy
$molecule
0 1
C 0.0 0.0 0.0
O 0.0 0.0 1.21
$end
$rem
EXCHANGE B3lyp
CIS_N_ROOTS 10
CIS_SINGLETS true
CIS_TRIPLETS true
RPA TRUE
BASIS 6-31+G*
XC_GRID 1
SOLVENT_METHOD pcm
$end
$pcm
Theory CPCM
Method SWIG
Solver Inversion
Radii Bondi
$end
$solvent
Dielectric 78.39
OpticalDielectric 1.777849
$end
7.3.5 Analytical Excited-State Hessian in TDDFT
To carry out vibrational frequency analysis of an excited state with TDDFT,[Liu and Liang(2011a), Liu and Liang(2011b)] an optimization of the excited-state geometry is always necessary. Like the vibrational frequency analysis of the ground state, the frequency analysis of the excited state should be also performed at a stationary point on the excited state potential surface. The $rem variable CIS_STATE_DERIV should be set to the excited state for which an optimization and frequency analysis is needed, in addition to the $rem keywords used for an excitation energy calculation.
Compared to the numerical differentiation method, the analytical calculation of geometrical second derivatives of the excitation energy needs much less time but much more memory. The computational cost is mainly consumed by the steps to solve both the CPSCF equations for the derivatives of molecular orbital coefficients C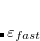 and the CP-TDDFT equations for the derivatives of the transition vectors, as well as to build the Hessian matrix. The memory usages for these steps scale as
and the CP-TDDFT equations for the derivatives of the transition vectors, as well as to build the Hessian matrix. The memory usages for these steps scale as  , where
, where  is the number of basis functions and m is the number of atoms. For large systems, it is thus essential to solve all the coupled-perturbed equations in segments. In this case, the $rem variable CPSCF_NSEG is always needed.
is the number of basis functions and m is the number of atoms. For large systems, it is thus essential to solve all the coupled-perturbed equations in segments. In this case, the $rem variable CPSCF_NSEG is always needed.
In the calculation of the analytical TDDFT excited-state Hessian, one has to evaluate a large number of energy-functional derivatives: the first-order to fourth-order functional derivatives with respect to the density variables as well as their derivatives with respect to the nuclear coordinates. Therefore, a very fine integration grid for DFT calculation should be adapted to guarantee the accuracy of the results.
Analytical TDDFT/C-PCM Hessian has been implemented in Q-Chem. Normal mode analysis for a system in solution can be performed with the frequency calculation by TDDFT/C-PCM method. The $rem and $pcm variables for the excited state calculation with TDDFT/C-PCM included in the vertical excitation energy example above are needed. When the properties of large systems are calculated, you must pay attention to the memory limit. At present, only a few exchange correlation functionals, including Slater+VWN, BLYP, B3LYP, are available for the analytical Hessian calculation.
Example 7.106 A B3LYP/6-31G* optimization in gas phase, followed by a frequency analysis for the first excited state of the peroxy radical
$molecule
0 2
C 1.004123 -0.180454 0.000000
O -0.246002 0.596152 0.000000
O -1.312366 -0.230256 0.000000
H 1.810765 0.567203 0.000000
H 1.036648 -0.805445 -0.904798
H 1.036648 -0.805445 0.904798
$end
$rem
JOBTYPE opt
EXCHANGE b3lyp
CIS_STATE_DERIV 1
BASIS 6-31G*
CIS_N_ROOTS 10
CIS_SINGLETS true
CIS_TRIPLETS false
XC_GRID 000075000302
RPA 0
$end
@@@
$molecule
read
$end
$rem
JOBTYPE freq
EXCHANGE b3lyp
CIS_STATE_DERIV 1
BASIS 6-31G*
CIS_N_ROOTS 10
CIS_SINGLETS true
CIS_TRIPLETS false
XC_GRID 000075000302
RPA 0
$end
Example 7.107 The optimization and Hessian calculation for low-lying excited state of 9-Fluorenone + 2 methanol in methanol solution using TDDFT/C-PCM
$molecule
0 1
6 -1.987249 0.699711 0.080583
6 -1.987187 -0.699537 -0.080519
6 -0.598049 -1.148932 -0.131299
6 0.282546 0.000160 0.000137
6 -0.598139 1.149219 0.131479
6 -0.319285 -2.505397 -0.285378
6 -1.386049 -3.395376 -0.388447
6 -2.743097 -2.962480 -0.339290
6 -3.049918 -1.628487 -0.186285
6 -3.050098 1.628566 0.186246
6 -2.743409 2.962563 0.339341
6 -1.386397 3.395575 0.388596
6 -0.319531 2.505713 0.285633
8 1.560568 0.000159 0.000209
1 0.703016 -2.862338 -0.324093
1 -1.184909 -4.453877 -0.510447
1 -3.533126 -3.698795 -0.423022
1 -4.079363 -1.292006 -0.147755
1 0.702729 2.862769 0.324437
1 -1.185378 4.454097 0.510608
1 -3.533492 3.698831 0.422983
1 -4.079503 1.291985 0.147594
8 3.323150 2.119222 0.125454
1 2.669309 1.389642 0.084386
6 3.666902 2.489396 -1.208239
1 4.397551 3.298444 -1.151310
1 4.116282 1.654650 -1.759486
1 2.795088 2.849337 -1.768206
1 2.669205 -1.389382 -0.084343
8 3.322989 -2.119006 -0.125620
6 3.666412 -2.489898 1.207974
1 4.396966 -3.299023 1.150789
1 4.115800 -1.655485 1.759730
1 2.794432 -2.850001 1.767593
$end
$rem
JOBTYPE OPT
EXCHANGE B3lyp
CIS_N_ROOTS 10
CIS_SINGLETS true
CIS_TRIPLETS true
CIS_STATE_DERIV 1 Lowest TDDFT state
RPA TRUE
BASIS 6-311G**
XC_GRID 000075000302
SOLVENT_METHOD pcm
$end
$pcm
Theory CPCM
Method SWIG
Solver Inversion
Radii Bondi
$end
$solvent
Dielectric 32.613
$end
@@@
$molecule
read
$end
$rem
JOBTYPE freq
EXCHANGE B3lyp
CIS_N_ROOTS 10
CIS_SINGLETS true
CIS_TRIPLETS true
RPA TRUE
CIS_STATE_DERIV 1 Lowest TDDFT state
BASIS 6-311G**
XC_GRID 000075000302
SOLVENT_METHOD pcm
MEM_STATIC 4000
MEM_TOTAL 24000
CPSCF_NSEG 3
$end
$pcm
Theory CPCM
Method SWIG
Solver Inversion
Radii Bondi
$end
$solvent
Dielectric 32.613
$end
7.3.6 Calculations of Spin-Orbit Couplings Between TDDFT States
Calculations of spin-orbit couplings (SOCs) for TDDFT states within the Tamm-Dancoff approximation or RPA (including TDHF and CIS states) is available. We employ the one-electron Breit Pauli Hamiltonian to calculate the SOC constant between TDDFT states.
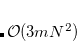 |
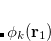 |
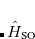 |
(7.16) |
where 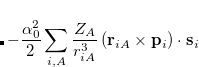 denotes electrons,
denotes electrons,  denotes nuclei,
denotes nuclei, 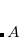 is the fine structure constant. Z
is the fine structure constant. Z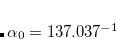 is the bare positive charge on nucleus A. In the second quantization representation, the spin-orbit Hamiltonian in different directions can be expressed as
is the bare positive charge on nucleus A. In the second quantization representation, the spin-orbit Hamiltonian in different directions can be expressed as
 |
|
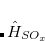 |
(7.17) | ||
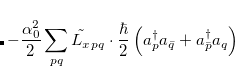 |
|
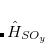 |
(7.18) | ||
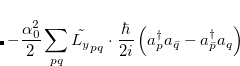 |
|
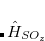 |
(7.19) |
where 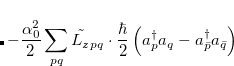 . The single-reference
. The single-reference 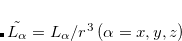 excited states (within the Tamm-Dancoff approximation) are given by
excited states (within the Tamm-Dancoff approximation) are given by
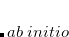 |
|
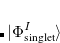 |
(7.20) | ||
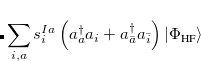 |
|
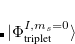 |
(7.21) | ||
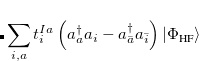 |
|
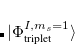 |
(7.22) | ||
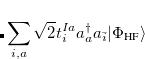 |
|
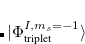 |
(7.23) |
where 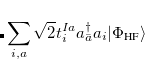 and
and 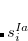 are singlet and triplet excitation coefficients of the
are singlet and triplet excitation coefficients of the 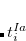 singlet or triplet state respectively, with the normalization
singlet or triplet state respectively, with the normalization 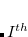 ;
; 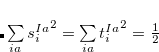 refers to the Hartree-Fock ground state. Thus the SOC constant from the singlet state to different triplet manifolds can be obtained as follows,
refers to the Hartree-Fock ground state. Thus the SOC constant from the singlet state to different triplet manifolds can be obtained as follows,
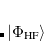 |
|
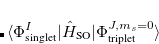 |
(7.24) | ||
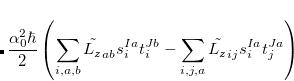 |
|
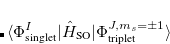 |
(7.25) | ||
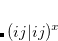 |
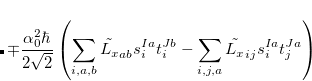 |
 |
(7.26) |
The SOC constant between different triplet manifolds can be obtained
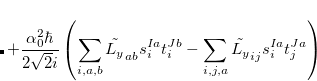 |
|
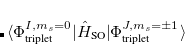 |
(7.27) | ||
|
|
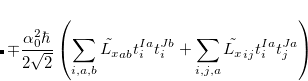 |
(7.28) | ||
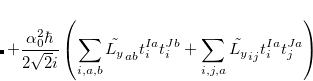 |
|
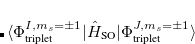 |
(7.29) |
Note that 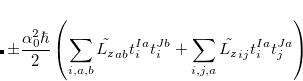 . The total (root-mean-square) spin-orbit coupling is given by
. The total (root-mean-square) spin-orbit coupling is given by
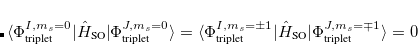 |
|
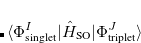 |
(7.30) | ||
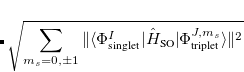 |
|
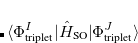 |
(7.31) |
For RPA states, the SOC constant can simply be obtained by replacing 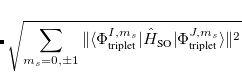 (
(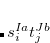 ) with
) with 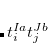 (
(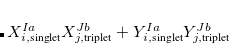 ) Setting the $rem variable CALC_SOC = TRUE will enable the SOC calculation for all calculated TDDFT states.
) Setting the $rem variable CALC_SOC = TRUE will enable the SOC calculation for all calculated TDDFT states.
CALC_SOC
Controls whether to calculate the SOC constants for EOM-CC, ADC, TDDFT/TDA and TDDFT.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform the SOC calculation.
TRUE
Perform the SOC calculation.
RECOMMENDATION:
None
Example 7.108 Calculation of SOCs for water molecule using TDDFT/B3LYP functional within the TDA.
$comment
This sample input calculates the spin-orbit coupling constants for water
between its ground state and its TDDFT/TDA excited triplets as well as the
coupling between its TDDFT/TDA singlets and triplets. Results are given in
cm-1.
$end
$molecule
0 1
H 0.000000 -0.115747 1.133769
H 0.000000 1.109931 -0.113383
O 0.000000 0.005817 -0.020386
$end
$rem
JOBTYPE sp
EXCHANGE b3lyp
BASIS 6-31G
CIS_N_ROOTS 4
CIS_CONVERGENCE 8
CORRELATION none
MAX_SCF_CYCLES 600
MAX_CIS_CYCLES 50
SCF_ALGORITHM diis
MEM_STATIC 300
MEM_TOTAL 2000
SYMMETRY false
SYM_IGNORE true
UNRESTRICTED false
CIS_SINGLETS true
CIS_TRIPLETS true
CALC_SOC true
SET_ITER 300
$end
7.3.7 Various TDDFT-Based Examples
Example 7.109 This example shows two jobs which request variants of time-dependent density functional theory calculations. The first job, using the default value of RPA = FALSE, performs TDDFT in the Tamm-Dancoff approximation (TDA). The second job, with RPA = TRUE performs a both TDA and full TDDFT calculations.
$comment
methyl peroxy radical
TDDFT/TDA and full TDDFT with 6-31+G*
$end
$molecule
0 2
C 1.00412 -0.18045 0.00000
O -0.24600 0.59615 0.00000
O -1.31237 -0.23026 0.00000
H 1.81077 0.56720 0.00000
H 1.03665 -0.80545 -0.90480
H 1.03665 -0.80545 0.90480
$end
$rem
EXCHANGE b
CORRELATION lyp
CIS_N_ROOTS 5
BASIS 6-31+G*
SCF_CONVERGENCE 7
$end
@@@
$molecule
read
$end
$rem
EXCHANGE b
CORRELATION lyp
CIS_N_ROOTS 5
RPA true
BASIS 6-31+G*
SCF_CONVERGENCE 7
$end
Example 7.110 This example shows a calculation of the excited states of a formamide-water complex within a reduced excitation space of the orbitals located on formamide
$comment
formamide-water TDDFT/TDA in reduced excitation space
$end
$molecule
0 1
H 1.13 0.49 -0.75
C 0.31 0.50 -0.03
N -0.28 -0.71 0.08
H -1.09 -0.75 0.67
H 0.23 -1.62 -0.22
O -0.21 1.51 0.47
O -2.69 1.94 -0.59
H -2.59 2.08 -1.53
H -1.83 1.63 -0.30
$end
$rem
EXCHANGE b3lyp
CIS_N_ROOTS 10
BASIS 6-31++G**
TRNSS TRUE
TRTYPE 1
CUTOCC 60
CUTVIR 40
CISTR_PRINT TRUE
$end
$solute
1
2
3
4
5
6
$end
Example 7.111 This example shows a calculation of the core-excited states at the oxygen 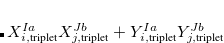 -edge of CO with a short-range corrected functional.
-edge of CO with a short-range corrected functional.
$comment
TDDFT with short-range corrected (SRC1) functional for the
oxygen K-edge of CO
$end
$molecule
0 1
C 0.000000 0.000000 -0.648906
O 0.000000 0.000000 0.486357
$end
$rem
EXCHANGE SRC1-R1
BASIS 6-311(2+,2+)G**
CIS_N_ROOTS 6
CIS_TRIPLETS false
TRNSS true
TRTYPE 3
N_SOL 1
$end
$solute
1
$end
Example 7.112 This example shows a calculation of the core-excited states at the phosphorus -edge with a short-range corrected functional.
$comment
TDDFT with short-range corrected (SRC2) functional for the
phosphorus K-edge of PH3
$end
$molecule
0 1
H 1.196206 0.000000 -0.469131
P 0.000000 0.000000 0.303157
H -0.598103 -1.035945 -0.469131
H -0.598103 1.035945 -0.469131
$end
$rem
EXCHANGE SRC2-R2
BASIS 6-311(2+,2+)G**
CIS_N_ROOTS 6
CIS_TRIPLETS false
TRNSS true
TRTYPE 3
N_SOL 1
$end
$solute
1
$end
Example 7.113 SF-TDDFT SP calculation of the 6 lowest states of the TMM diradical using recommended 50-50 functional
$molecule
0 3
C
C 1 CC1
C 1 CC2 2 A2
C 1 CC2 2 A2 3 180.0
H 2 C2H 1 C2CH 3 0.0
H 2 C2H 1 C2CH 4 0.0
H 3 C3Hu 1 C3CHu 2 0.0
H 3 C3Hd 1 C3CHd 4 0.0
H 4 C3Hu 1 C3CHu 2 0.0
H 4 C3Hd 1 C3CHd 3 0.0
CC1 = 1.35
CC2 = 1.47
C2H = 1.083
C3Hu = 1.08
C3Hd = 1.08
C2CH = 121.2
C3CHu = 120.3
C3CHd = 121.3
A2 = 121.0
$end
$rem
EXCHANGE gen
BASIS 6-31G*
SCF_GUESS core
SCF_CONVERGENCE 10
MAX_SCF_CYCLES 100
SPIN_FLIP 1
CIS_N_ROOTS 6
CIS_CONVERGENCE 10
MAX_CIS_CYCLES 100
$end
$xc_functional
X HF 0.50
X S 0.08
X B 0.42
C VWN 0.19
C LYP 0.81
$end
Example 7.114 SF-DFT with non-collinear exchange-correlation functional for low-lying states of 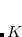
$comment
Non-collinear SF-DFT calculation for CH2 at 3B1 state geometry from
EOM-CCSD(fT) calculation
$end
$molecule
0 3
C
H 1 rCH
H 1 rCH 2 HCH
rCH = 1.0775
HCH = 133.29
$end
$rem
EXCHANGE PBE0
BASIS cc-pVTZ
SPIN_FLIP 1
WANG_ZIEGLER_KERNEL TRUE
SCF_CONVERGENCE 10
CIS_N_ROOTS 6
CIS_CONVERGENCE 10
$end