4.5 Converging SCF Calculations
4.5.1 Introduction
As for any numerical optimization procedure, the rate of convergence of the SCF procedure is dependent on the initial guess and on the algorithm used to step towards the stationary point. Q-Chem features a number of SCF optimization algorithms which can be selected via the $rem variable SCF_ALGORITHM, including:
Methods that are based on extrapolation/interpolation:
The highly successful DIIS procedures. These are the default (except for restricted open-shell SCF calculations) and are available for all orbital types (see Section 4.5.3).
ADIIS (the Accelerated DIIS algorithm developed by Hu et al.,[Hu and Yang(2010)] available for R and U only).
Methods that make use of orbital gradient:
Direct Minimization (DM), which has been re-implemented as simple steepest descent with line search, and is available for all orbital types. DM can be invoked after a few DIIS iterations.
Geometric Direct Minimization (GDM) which is an improved and highly robust version of DM and is the recommended fall-back when DIIS fails. Like DM, It can also be invoked after a few iterations with DIIS to improve the initial guess. GDM is the default algorithm for restricted open-shell SCF calculations and is available for all orbital types (see Section 4.5.4).
GDM_LS (it is essentially a preconditioned (using orbital energy differences as the preconditioner) L-BFGS algorithm with line search, available for R, U, RO and OS_RO).
Methods that require orbital Hessian:
NEWTON_CG/NEWTON_MINRES (solve
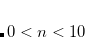 for the update direction with CG/MINRES solvers).
for the update direction with CG/MINRES solvers). SF_NEWTON_CG (the “saddle-free" version of NEWTON_CG).
The analytical orbital Hessian is available for R/U/RO/G/CR unless special density functionals (e.g. those containing VV10) are used, while the use of finite-difference Hessian is available for all orbital types by setting FD_MAT_VEC_PROD = TRUE.
In addition to these algorithms, there is also the maximum overlap method (MOM) which ensures that DIIS always occupies a continuous set of orbitals and does not oscillate between different occupancies. MOM can also be used to obtain higher-energy solutions of the SCF equations (see Section 7.4). The relaxed constraint algorithm (RCA), which guarantees that the energy goes down at every step, is also available via the old SCF code (set GEN_SCFMAN = FALSE). Nevertheless, the performance of the ADIIS algorithm should be similar to it.
Since the code in GEN_SCFMAN is highly modular, the availability of different SCF algorithms to different SCF (orbital) types is largely extended in general. For example, the old ROSCF implementation requires the use of the GWH guess and the GDM algorithm exclusively. Such a limitation has been eliminated in GEN_SCFMAN based RO calculations.
4.5.2 Basic Convergence Control Options
See also more detailed options in the following sections, and note that the SCF convergence criterion and the integral threshold must be set in a compatible manner, (this usually means THRESH should be set to at least 3 higher than SCF_CONVERGENCE).
MAX_SCF_CYCLES
Controls the maximum number of SCF iterations permitted.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:

User-selected.
RECOMMENDATION:
Increase for slowly converging systems such as those containing transition metals.
SCF_ALGORITHM
Algorithm used for converging the SCF.
TYPE:
STRING
DEFAULT:
DIIS
Pulay DIIS.
OPTIONS:
DIIS
Pulay DIIS.
DM
Direct minimizer.
DIIS_DM
Uses DIIS initially, switching to direct minimizer for later iterations
(See THRESH_DIIS_SWITCH, MAX_DIIS_CYCLES).
DIIS_GDM
Use DIIS and then later switch to geometric direct minimization
(See THRESH_DIIS_SWITCH, MAX_DIIS_CYCLES).
GDM
Geometric Direct Minimization.
RCA
Relaxed constraint algorithm
RCA_DIIS
Use RCA initially, switching to DIIS for later iterations (see
THRESH_RCA_SWITCH and MAX_RCA_CYCLES described
later in this chapter)
ROOTHAAN
Roothaan repeated diagonalization.
RECOMMENDATION:
Use DIIS unless performing a restricted open-shell calculation, in which case GDM is recommended. If DIIS fails to find a reasonable approximate solution in the initial iterations, RCA_DIIS is the recommended fallback option. If DIIS approaches the correct solution but fails to finally converge, DIIS_GDM is the recommended fallback.
SCF_CONVERGENCE
SCF is considered converged when the wave function error is less that
. Adjust the value of THRESH at the same time. Note as of Q-Chem 3.0 the DIIS error is measured by the maximum error rather than the RMS error.
TYPE:
INTEGER
DEFAULT:
5
For single point energy calculations.
7
For geometry optimizations and vibrational analysis.
8
For SSG calculations, see Chapter 6.
OPTIONS:
Corresponding to
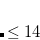
RECOMMENDATION:
Tighter criteria for geometry optimization and vibration analysis. Larger values provide more significant figures, at greater computational cost.
In some cases besides the total SCF energy, one needs its separate energy components, like kinetic energy, exchange energy, correlation energy, etc. The values of these components are printed at each SCF cycle if one specifies SCF_PRINT = 1 in the input.
4.5.3 Direct Inversion in the Iterative Subspace (DIIS)
The SCF implementation of the Direct Inversion in the Iterative Subspace (DIIS) method[Pulay(1980), Pulay(1982)] uses the property of an SCF solution that requires the density matrix to commute with the Fock matrix:
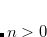 |
(4.33) |
During the SCF cycles, prior to achieving self-consistency, it is therefore possible to define an error vector e , which is non-zero except at convergence:
, which is non-zero except at convergence:
 |
(4.34) |
Here 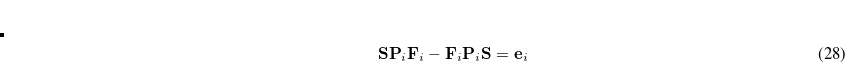 is obtained by diagonalizing
is obtained by diagonalizing 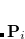 , and
, and
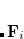 |
(4.35) |
The DIIS coefficients 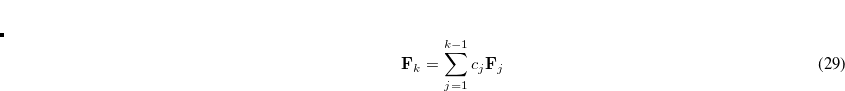 , are obtained by a least-squares constrained minimization of the error vectors, viz
, are obtained by a least-squares constrained minimization of the error vectors, viz
 |
(4.36) |
where the constraint 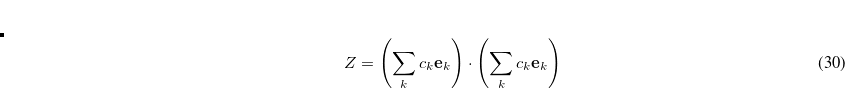 is imposed to yield a set of linear equations, of dimension
is imposed to yield a set of linear equations, of dimension 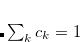 :
:
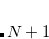 |
(4.37) |
Convergence criteria require the largest element of the  th error vector to be below a cutoff threshold, usually
th error vector to be below a cutoff threshold, usually 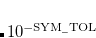 a.u. for single point energies, but often increased to
a.u. for single point energies, but often increased to 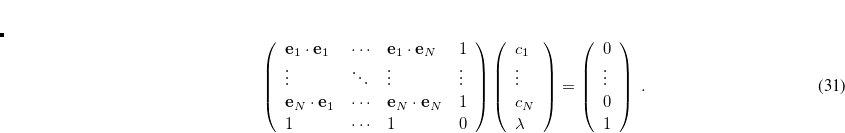 a.u. for optimizations and frequency calculations.
a.u. for optimizations and frequency calculations.
The rate of convergence may be improved by restricting the number of previous Fock matrices (size of the DIIS subspace, $rem variable DIIS_SUBSPACE_SIZE) used for determining the DIIS coefficients:
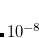 |
(4.38) |
where 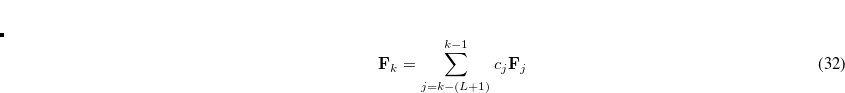 is the size of the DIIS subspace. As the Fock matrix nears self-consistency, the linear matrix equations in Eq. (4.37) tend to become severely ill-conditioned and it is often necessary to reset the DIIS subspace (this is automatically carried out by the program).
is the size of the DIIS subspace. As the Fock matrix nears self-consistency, the linear matrix equations in Eq. (4.37) tend to become severely ill-conditioned and it is often necessary to reset the DIIS subspace (this is automatically carried out by the program).
Finally, on a practical note, we observe that DIIS has a tendency to converge to global minima rather than local minima when employed for SCF calculations. This seems to be because only at convergence is the density matrix in the DIIS iterations idempotent. On the way to convergence, one is not on the true energy surface, and this seems to permit DIIS to “tunnel” through barriers in wave function space. This is usually a desirable property, and is the motivation for the options that permit initial DIIS iterations before switching to direct minimization to converge to the minimum in difficult cases.
The following $rem variables permit some customization of the DIIS iterations:
DIIS_SUBSPACE_SIZE
Controls the size of the DIIS and/or RCA subspace during the SCF.
TYPE:
INTEGER
DEFAULT:
15
OPTIONS:
User-defined
RECOMMENDATION:
None
DIIS_PRINT
Controls the output from DIIS SCF optimization.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Minimal print out.
1
Chosen method and DIIS coefficients and solutions.
2
Level 1 plus changes in multipole moments.
3
Level 2 plus Multipole moments.
4
Level 3 plus extrapolated Fock matrices.
RECOMMENDATION:
Use the default
Note: In Q-Chem 3.0 the DIIS error is determined by the maximum error rather than the RMS error. For backward compatibility the RMS error can be forced by using the following $rem
DIIS_ERR_RMS
Changes the DIIS convergence metric from the maximum to the RMS error.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE, FALSE
RECOMMENDATION:
Use the default, the maximum error provides a more reliable criterion.
DIIS_SEPARATE_ERRVEC
Control optimization of DIIS error vector in unrestricted calculations.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use a combined
and
error vector.
OPTIONS:
FALSE
Use a combined
TRUE
Use separate error vectors for the
RECOMMENDATION:
When using DIIS in Q-Chem a convenient optimization for unrestricted calculations is to sum the
4.5.4 Geometric Direct Minimization (GDM)
Troy Van Voorhis, working at Berkeley with Martin Head-Gordon, has developed a novel direct minimization method that is extremely robust, and at the same time is only slightly less efficient than DIIS. This method is called geometric direct minimization (GDM) because it takes steps in an orbital rotation space that correspond properly to the hyper-spherical geometry of that space. In other words, rotations are variables that describe a space which is curved like a many-dimensional sphere. Just like the optimum flight paths for airplanes are not straight lines but great circles, so too are the optimum steps in orbital rotation space. GDM takes this correctly into account, which is the origin of its efficiency and its robustness. For full details, we refer the reader to Ref. VanVoorhis:2002a. GDM is a good alternative to DIIS for SCF jobs that exhibit convergence difficulties with DIIS.
Recently, Barry Dunietz, also working at Berkeley with Martin Head-Gordon, has extended the GDM approach to restricted open-shell SCF calculations. Their results indicate that GDM is much more efficient than the older direct minimization method (DM).
In section 4.5.3, we discussed the fact that DIIS can efficiently head towards the global SCF minimum in the early iterations. This can be true even if DIIS fails to converge in later iterations. For this reason, a hybrid scheme has been implemented which uses the DIIS minimization procedure to achieve convergence to an intermediate cutoff threshold. Thereafter, the geometric direct minimization algorithm is used. This scheme combines the strengths of the two methods quite nicely: the ability of DIIS to recover from initial guesses that may not be close to the global minimum, and the ability of GDM to robustly converge to a local minimum, even when the local surface topology is challenging for DIIS. This is the recommended procedure with which to invoke GDM (i.e., setting SCF_ALGORITHM = DIIS_GDM). This hybrid procedure is also compatible with the SAD guess, while GDM itself is not, because it requires an initial guess set of orbitals. If one wishes to disturb the initial guess as little as possible before switching on GDM, one should additionally specify MAX_DIIS_CYCLES = 1 to obtain only a single Roothaan step (which also serves up a properly orthogonalized set of orbitals).
$rem options relevant to GDM are SCF_ALGORITHM which should be set to either GDM or DIIS_GDM and the following:
MAX_DIIS_CYCLES
The maximum number of DIIS iterations before switching to (geometric) direct minimization when SCF_ALGORITHM is DIIS_GDM or DIIS_DM. See also THRESH_DIIS_SWITCH.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
1
Only a single Roothaan step before switching to (G)DM
RECOMMENDATION:
None
THRESH_DIIS_SWITCH
The threshold for switching between DIIS extrapolation and direct minimization of the SCF energy is
when SCF_ALGORITHM is DIIS_GDM or DIIS_DM. See also MAX_DIIS_CYCLES
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
User-defined.
RECOMMENDATION:
None
4.5.5 Direct Minimization (DM)
Direct minimization (DM) is a less sophisticated forerunner of the geometric direct minimization (GDM) method discussed in the previous section. DM does not properly step along great circles in the hyper-spherical space of orbital rotations, and therefore converges less rapidly and less robustly than GDM, in general. It is retained for legacy purposes, and because it is at present the only method available for restricted open shell (RO) SCF calculations in Q-Chem. In general, the input options are the same as for GDM, with the exception of the specification of SCF_ALGORITHM, which can be either DIIS_DM (recommended) or DM.
PSEUDO_CANONICAL
When SCF_ALGORITHM = DM, this controls the way the initial step, and steps after subspace resets are taken.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Use Roothaan steps when (re)initializing
TRUE
Use a steepest descent step when (re)initializing
RECOMMENDATION:
The default is usually more efficient, but choosing TRUE sometimes avoids problems with orbital reordering.
4.5.6 Maximum Overlap Method (MOM)
In general, the DIIS procedure is remarkably successful. One difficulty that is occasionally encountered is the problem of an SCF that occupies two different sets of orbitals on alternating iterations, and therefore oscillates and fails to converge. This can be overcome by choosing orbital occupancies that maximize the overlap of the new occupied orbitals with the set previously occupied. Q-Chem contains the maximum overlap method (MOM),[Gilbert et al.(2008)Gilbert, Besley, and Gill] developed by Andrew Gilbert and Peter Gill.
MOM is therefore is a useful adjunct to DIIS in convergence problems involving flipping of orbital occupancies. It is controlled by the $rem variable MOM_START, which specifies the SCF iteration on which the MOM procedure is first enabled. There are two strategies that are useful in setting a value for MOM_START. To help maintain an initial configuration it should be set to start on the first cycle. On the other hand, to assist convergence it should come on later to avoid holding on to an initial configuration that may be far from the converged one.
The MOM-related $rem variables in full are the following:.
MOM_PRINT
Switches printing on within the MOM procedure.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Printing is turned off
TRUE
Printing is turned on.
RECOMMENDATION:
None
MOM_START
Determines when MOM is switched on to stabilize DIIS iterations.
TYPE:
INTEGER
DEFAULT:
0 (FALSE)
OPTIONS:
0 (FALSE)
MOM is not used
MOM begins on cycle
RECOMMENDATION:
Set to 1 if preservation of initial orbitals is desired. If MOM is to be used to aid convergence, an SCF without MOM should be run to determine when the SCF starts oscillating. MOM should be set to start just before the oscillations.
MOM_METHOD
Determines the target orbitals with which to maximize the overlap on each SCF cycle.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
3
Maximize overlap with the orbitals from the previous SCF cycle.
13
Maximize overlap with the initial guess orbitals.
RECOMMENDATION:
If appropriate guess orbitals can be obtained, then MOM_METHOD = 13 can provide more reliable convergence to the desired solution.
4.5.7 Relaxed Constraint Algorithm (RCA)
The relaxed constraint algorithm (RCA) is an ingenious and simple means of minimizing the SCF energy that is particularly effective in cases where the initial guess is poor. The latter is true, for example, when employing a user-specified basis (when the “core” or GWH guess must be employed) or when near-degeneracy effects imply that the initial guess will likely occupy the wrong orbitals relative to the desired converged solution.
Briefly, RCA begins with the SCF problem as a constrained minimization of the energy as a function of the density matrix, 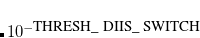 .[Cancès and Le Bris(2000), Cancès(2001)] The constraint is that the density matrix be idempotent,
.[Cancès and Le Bris(2000), Cancès(2001)] The constraint is that the density matrix be idempotent, 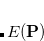 , which basically forces the occupation numbers to be either zero or one. The fundamental realization of RCA is that this constraint can be relaxed to allow sub-idempotent density matrices,
, which basically forces the occupation numbers to be either zero or one. The fundamental realization of RCA is that this constraint can be relaxed to allow sub-idempotent density matrices, 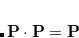 . This condition forces the occupation numbers to be between zero and one. Physically, we expect that any state with fractional occupations can lower its energy by moving electrons from higher energy orbitals to lower ones. Thus, if we solve for the minimum of subject to the relaxed sub-idempotent constraint, we expect that the ultimate solution will nonetheless be idempotent. In fact, for Hartree-Fock this can be rigorously proven. For density functional theory, it is possible that the minimum will have fractional occupation numbers but these occupations have a physical interpretation in terms of ensemble DFT. The reason the relaxed constraint is easier to deal with is that it is easy to prove that a linear combination of sub-idempotent matrices is also sub-idempotent as long as the linear coefficients are between zero and one. By exploiting this property, convergence can be accelerated in a way that guarantees the energy will go down at every step.
. This condition forces the occupation numbers to be between zero and one. Physically, we expect that any state with fractional occupations can lower its energy by moving electrons from higher energy orbitals to lower ones. Thus, if we solve for the minimum of subject to the relaxed sub-idempotent constraint, we expect that the ultimate solution will nonetheless be idempotent. In fact, for Hartree-Fock this can be rigorously proven. For density functional theory, it is possible that the minimum will have fractional occupation numbers but these occupations have a physical interpretation in terms of ensemble DFT. The reason the relaxed constraint is easier to deal with is that it is easy to prove that a linear combination of sub-idempotent matrices is also sub-idempotent as long as the linear coefficients are between zero and one. By exploiting this property, convergence can be accelerated in a way that guarantees the energy will go down at every step.
The implementation of RCA in Q-Chem closely follows the “Energy DIIS” implementation of the RCA algorithm.[Kudin et al.(2002)Kudin, Scuseria, and Cancès] Here, the current density matrix is written as a linear combination of the previous density matrices:
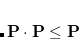 |
(4.39) |
To a very good approximation (exact for Hartree-Fock) the energy for 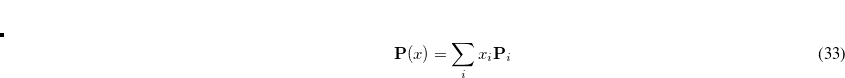 can be written as a quadratic function of x:
can be written as a quadratic function of x:
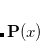 |
(4.40) |
At each iteration, 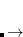 is chosen to minimize
is chosen to minimize  subject to the constraint that all of the
subject to the constraint that all of the 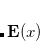 are between zero and one. The Fock matrix for is further written as a linear combination of the previous Fock matrices,
are between zero and one. The Fock matrix for is further written as a linear combination of the previous Fock matrices,
 |
(4.41) |
where 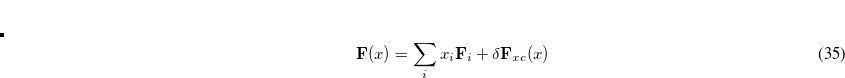 denotes a (usually quite small) change in the exchange-correlation part that is computed once has been determined. We note that this extrapolation is very similar to that used by DIIS. However, this procedure is guaranteed to reduce the energy at every iteration, unlike DIIS.
denotes a (usually quite small) change in the exchange-correlation part that is computed once has been determined. We note that this extrapolation is very similar to that used by DIIS. However, this procedure is guaranteed to reduce the energy at every iteration, unlike DIIS.
In practice, the RCA approach is ideally suited to difficult convergence situations because it is immune to the erratic orbital swapping that can occur in DIIS. On the other hand, RCA appears to perform relatively poorly near convergence, requiring a relatively large number of steps to improve the precision of a good approximate solution. It is thus advantageous in many cases to run RCA for the initial steps and then switch to DIIS either after some specified number of iterations or after some target convergence threshold has been reached. Finally, note that by its nature RCA considers the energy as a function of the density matrix. As a result, it cannot be applied to restricted open shell calculations which are explicitly orbital-based. Note: RCA interacts poorly with INCDFT, so INCDFT is disabled by default when an RCA or RCA_DIIS calculation is requested. To enable INCDFT with such a calculation, set INCDFT = 2 in the $rem section. RCA may also have poor interactions with incremental Fock builds; if RCA fails to converge, setting INCFOCK = FALSE may improve convergence in some cases.
Job-control variables for RCA are listed below, and an example input can be found in Section 4.5.11.
RCA_PRINT
Controls the output from RCA SCF optimizations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No print out
1
RCA summary information
2
Level 1 plus RCA coefficients
3
Level 2 plus RCA iteration details
RECOMMENDATION:
None
MAX_RCA_CYCLES
The maximum number of RCA iterations before switching to DIIS when SCF_ALGORITHM is RCA_DIIS.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
N
N RCA iterations before switching to DIIS
RECOMMENDATION:
None
THRESH_RCA_SWITCH
The threshold for switching between RCA and DIIS when SCF_ALGORITHM is RCA_DIIS.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
N
Algorithm changes from RCA to DIIS when Error is less than
.
RECOMMENDATION:
None
4.5.8 User-Customized Hybrid SCF Algorithm
It is often the case that a single algorithm is not able to guarantee SCF convergence. Meanwhile, some SCF algorithms (e.g. ADIIS) can accelerate convergence at the beginning of an SCF calculation but becomes less efficient near the convergence. While a few hybrid algorithms (DIIS_GDM, RCA_DIIS) have been enabled in Q-Chem’s original SCF implementation, in GEN_SCFMAN, we seek for a more flexible setup for the use of multiple SCF algorithms so that users can have a more precise control on the SCF procedure. With the current implementation, at most four distinct algorithms (usually more than enough) can be employed in one single SCF calculation based on GEN_SCFMAN, and the basic job control is as follows:
GEN_SCFMAN_HYBRID_ALGO
Use multiple algorithms in an SCF calculation based on GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Use a single SCF algorithm (given by SCF_ALGORITHM).
TRUE
Use multiple SCF algorithms (to be specified).
RECOMMENDATION:
Set it to TRUE when the use of more than one algorithm is desired.
GEN_SCFMAN_ALGO_1
The first algorithm to be used in a hybrid-algorithm calculation.
TYPE:
STRING
DEFAULT:
0
OPTIONS:
All the available SCF_ALGORITHM options, including the GEN_SCFMAN additions (Section 4.3.1).
RECOMMENDATION:
None
GEN_SCFMAN_ITER_1
Maximum number of iterations given to the first algorithm. If used up, switch to the next algorithm.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
User-defined
RECOMMENDATION:
None
GEN_SCFMAN_CONV_1
The convergence criterion given to the first algorithm. If reached, switch to the next algorithm.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
10
RECOMMENDATION:
None
Note: $rem variables GEN_SCFMAN_ALGO_X, GEN_SCFMAN_ITER_X, GEN_SCFMAN_CONV_X (X = 2, 3, 4) are defined and used in a similar way.
Example 4.29 B3LYP/3-21G calculation for a cadmium-imidazole complex using the ADIIS-DIIS algorithm (an example from Ref. Hu:2010). Due to the poor quality of the CORE guess, using a single algorithm such as DIIS or GDM fails to converge.
$molecule
2 1
Cd 0.000000 0.000000 0.000000
N 0.000000 0.000000 -2.260001
N -0.685444 0.000000 -4.348035
C 0.676053 0.000000 -4.385069
C 1.085240 0.000000 -3.091231
C -1.044752 0.000000 -3.060220
H 1.231530 0.000000 -5.300759
H 2.088641 0.000000 -2.711077
H -2.068750 0.000000 -2.726515
H -1.313170 0.000000 -5.174718
$end
$rem
JOBTYPE SP
EXCHANGE B3LYP
BASIS 3-21g
UNRESTRICTED FALSE
SYMMETRY FALSE
SYM_IGNORE TRUE
THRESH 14
SCF_GUESS CORE
GEN_SCFMAN TRUE
GEN_SCFMAN_HYBRID_ALGO TRUE
GEN_SCFMAN_ALGO_1 ADIIS
GEN_SCFMAN_CONV_1 3 !switch to DIIS when error < 1E-3
GEN_SCFMAN_ITER_1 50
GEN_SCFMAN_ALGO_2 DIIS
GEN_SCFMAN_CONV_2 8
GEN_SCFMAN_ITER_2 50
$end
4.5.9 Internal Stability Analysis and Automated Correction for Energy Minima
At convergence, the SCF energy will be at a stationary point with respect to changes in the MO coefficients. However, this stationary point is not guaranteed to be an energy minimum, and in cases where it is not, the wave function is said to be unstable. Even if the wave function is at a minimum, this minimum may be an artifact of the constraints placed on the form of the wave function. For example, an unrestricted calculation will usually give a lower energy than the corresponding restricted calculation, and this can give rise to an RHF  UHF instability.
UHF instability.
Based on our experience, even for very simple data set such as the G2 atomization energies,[Curtiss et al.(1997)Curtiss, Raghavachari, Redfern, and Pople] using the default algorithm (DIIS) produces unstable solutions for several species (even for single atoms with some density functionals). In such cases, failure to check the internal stability of SCF solutions can result in flawed benchmark results. Although in general the use of gradient-based algorithms such as GDM is more likely to locate the true minimum, it still cannot entirely eliminate the possibility of finding an unstable solution.
To understand what instabilities can occur, it is useful to consider the most general form possible for the spin orbitals:
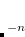 |
(4.42) |
Here, 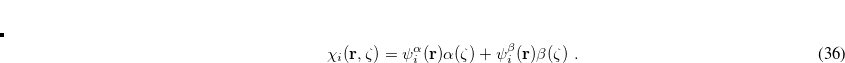 and
and 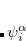 are complex-valued functions of the Cartesian coordinates
are complex-valued functions of the Cartesian coordinates 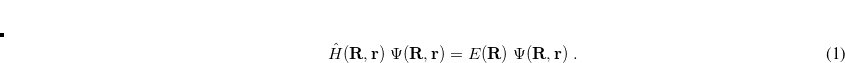 , and and are spin eigenfunctions of the spin-variable
, and and are spin eigenfunctions of the spin-variable  . The first constraint that is almost universally applied is to assume the spin orbitals depend only on one or other of the spin-functions or . Thus, the spin-functions take the form
. The first constraint that is almost universally applied is to assume the spin orbitals depend only on one or other of the spin-functions or . Thus, the spin-functions take the form
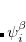 |
(4.43) |
In addition, most SCF calculations use real functions, and this places an additional constraint on the form of the wave function. If there exists a complex solution to the SCF equations that has a lower energy, the wave function exhibits a real complex instability. The final constraint that is commonly placed on the spin-functions is that 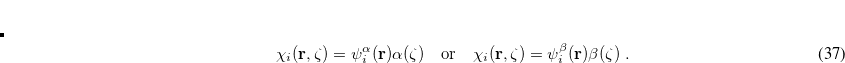 , i.e., that the spatial parts of the spin-up and spin-down orbitals are the same. This gives the familiar restricted formalism and can lead to an RHF UHF instability as mentioned above. Further details about the possible instabilities can be found in Ref. Seeger:1977.
, i.e., that the spatial parts of the spin-up and spin-down orbitals are the same. This gives the familiar restricted formalism and can lead to an RHF UHF instability as mentioned above. Further details about the possible instabilities can be found in Ref. Seeger:1977.
Wave function instabilities can arise for several reasons, but frequently occur if
There exists a singlet diradical at a lower energy then the closed-shell singlet state.
There exists a triplet state at a lower energy than the lowest singlet state.
There are multiple solutions to the SCF equations, and the calculation has not found the lowest energy solution.
Q-Chem’s previous stability analysis package suffered from the following limitations:
It is only available for restricted (close-shell) and unrestricted SCF calculations.
It requires the analytical orbital Hessian of the wave function energy.
The calculation terminates after the corrected MOs are generated, and a second job is needed to read in these orbitals and run another SCF calculation.
The implementation of internal stability analysis in GEN_SCFMAN overcomes almost all these shortcomings. Its availability has been extended to all the implemented orbital types. As in the old code, when the analytical Hessian of the given orbital type and theory (e.g. RO/B3LYP) is available, it computes matrix-vector products analytically for the Davidson algorithm.[Davidson(1975)] If the analytical Hessian is not available, users can still run stability analysis by using the finite-difference matrix-vector product technique developed by Sharada et al.,[Sharada et al.(2015)Sharada, Stück, Sundstrom, Bell, and Head-Gordon] which requires the gradient (related to the Fock matrix) only:
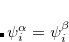 |
(4.44) |
where 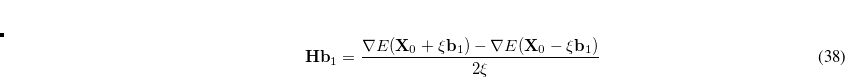 is the Hessian matrix,
is the Hessian matrix, 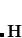 is a trial vector,
is a trial vector, 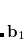 stands for the current stationary point, and
stands for the current stationary point, and 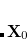 is the finite step size. With this method, internal stability analysis is available for all the implemented orbital types in GEN_SCFMAN. It should be noted that since the second derivative of NLC functionals such as VV10 is not available in Q-Chem, this finite-difference method will be used by default for the evaluation of Hessian-vector products.
is the finite step size. With this method, internal stability analysis is available for all the implemented orbital types in GEN_SCFMAN. It should be noted that since the second derivative of NLC functionals such as VV10 is not available in Q-Chem, this finite-difference method will be used by default for the evaluation of Hessian-vector products.
GEN_SCFMAN allows multiple SCF calculations and stability analyses to be performed in a single job so that it can make use of the corrected MOs and locate the true minimum automatically. The MOs are displaced along the direction of the lowest-energy eigenvector (with line search) if an SCF solution is found to be unstable. A new SCF calculation that reads in these corrected MOs as initial guess will be launched automatically if INTERNAL_STABILITY_ITER > 0. Such macro-loops will keep going until a stable solution is reached.
Note: The stability analysis package can be used to analyze both HF and DFT wave functions.
4.5.9.1 Job Control
INTERNAL_STABILITY
Perform internal stability analysis in GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform internal stability analysis after convergence.
TRUE
Perform internal stability analysis and generate the corrected MOs.
RECOMMENDATION:
Turn it on when the SCF solution is prone to unstable solutions, especially for open-shell species.
FD_MAT_VEC_PROD
Compute Hessian-vector product using the finite difference technique. |
TYPE:
BOOLEAN |
DEFAULT:
FALSE (TRUE when the employed functional contains NLC) |
OPTIONS:
FALSE |
Compute Hessian-vector product analytically. |
TRUE |
Use finite difference to compute Hessian-vector product. |
RECOMMENDATION:
Set it to TRUE when analytical Hessian is not available. |
Note: |
only the NLC part will be computed with finite difference.
INTERNAL_STABILITY_ITER
Maximum number of new SCF calculations permitted after the first stability analysis is performed.
TYPE:
INTEGER
DEFAULT:
0 (automatically set to 1 if INTERNAL_STABILITY = TRUE)
OPTIONS:
RECOMMENDATION:
Give a larger number if 1 is not enough (still unstable).
INTERNAL_STABILITY_DAVIDSON_ITER
Maximum number of Davidson iterations allowed in one stability analysis.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
Perform up to
RECOMMENDATION:
Use the default.
INTERNAL_STABILITY_CONV
Convergence criterion for the Davidson solver (for the lowest eigenvalues).
TYPE:
INTEGER
DEFAULT:
4 (3 when FD_MAT_ON_VECS = TRUE)
OPTIONS:
Terminate Davidson iterations when the norm of the residual vector is below 10
RECOMMENDATION:
Use the default.
INTERNAL_STABILITY_ROOTS
Number of lowest Hessian eigenvalues to solve for.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
Solve for
RECOMMENDATION:
Use the default.
Example 4.30 Unrestricted SCF calculation of triplet B using B97M-V/6-31g with the GDM algorithm. A displacement is performed when the first solution is characterized as a saddle point, and the second SCF gives a stable solution.
using B97M-V/6-31g with the GDM algorithm. A displacement is performed when the first solution is characterized as a saddle point, and the second SCF gives a stable solution.
$molecule
0 3
b
b 1 R
R = 1.587553
$end
$rem
JOBTYPE sp
METHOD b97m-v
BASIS 6-31g
UNRESTRICTED true
THRESH 14
SYMMETRY false
SYM_IGNORE true
SCF_FINAL_PRINT 1
SCF_ALGORITHM gdm
SCF_CONVERGENCE 8
INTERNAL_STABILITY true !turn on internal stability analysis
FD_MAT_VEC_PROD false !use finite-diff for the vv10 part only
$end
4.5.10 Small-Gap Systems
SCF calculations for systems with zero or small HOMO-LUMO gap (such as metals) can exhibit very slow convergence or may even fail to converge. This problem arises because the energetic ordering of orbitals and states can switch during the SCF optimization leading to discontinuities in the optimization. Using fractional MO occupation numbers can improve the convergence for small-gap systems. In this approach, the occupation numbers of MOs around the Fermi level are allowed to assume non-integer values. This “occupation smearing” allows one to include multiple electron configurations in the same optimization, which improves the stability of the optimization.
We follow the pseudo-Fractional Occupation Number (pFON) method of Rabuck and Scuseria[Rabuck and Scuseria(1999)] that scales the MO occupation used to construct the AO density:
 |
(4.45) |
For a conventional (integer occupation number) SCF run, the occupation number 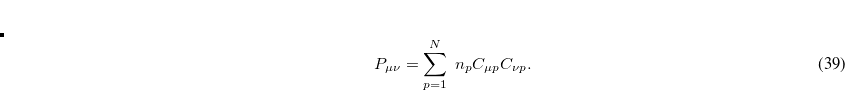 is either one (occupied) or zero (virtual). In pFON, the occupation numbers are following a Fermi-Dirac distribution,
is either one (occupied) or zero (virtual). In pFON, the occupation numbers are following a Fermi-Dirac distribution,
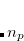 |
(4.46) |
where 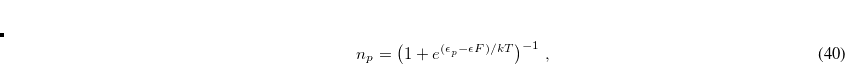 is the respective orbital energy and
is the respective orbital energy and 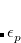 the Boltzmann constant and temperature, respectively. The Fermi energy
the Boltzmann constant and temperature, respectively. The Fermi energy 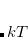 is set to
is set to 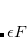 in our implementation. To ensure conservation of the total number of electrons, the pFON approach re-scales the occupation numbers so that
in our implementation. To ensure conservation of the total number of electrons, the pFON approach re-scales the occupation numbers so that 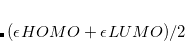 .
.
There are several parameters to control the electronic temperature 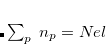 throughout a pFON SCF run. The temperature can either be held constant at finite temperature (
throughout a pFON SCF run. The temperature can either be held constant at finite temperature (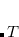 =
= 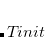 ), or the system can be cooled from a higher temperature down to the final temperature. So far, no zero-temperature extrapolation has been implemented.
), or the system can be cooled from a higher temperature down to the final temperature. So far, no zero-temperature extrapolation has been implemented.
OCCUPATIONS
Activates pFON calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Integer occupation numbers
1
Not yet implemented
2
Pseudo-fractional occupation numbers (pFON)
RECOMMENDATION:
Use pFON to improve convergence for small-gap systems.
FON_T_START
Initial electronic temperature (in K) for FON calculation.
TYPE:
INTEGER
DEFAULT:
1000
OPTIONS:
Any desired initial temperature.
RECOMMENDATION:
Pick the temperature to either reproduce experimental conditions (e.g. room temperature) or as low as possible to approach zero-temperature.
FON_T_END
Final electronic temperature for FON calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Any desired final temperature.
RECOMMENDATION:
Pick the temperature to either reproduce experimental conditions (e.g. room temperature) or as low as possible to approach zero-temperature.
FON_NORB
Number of orbitals above and below the Fermi level that are allowed to have fractional occupancies.
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
n
number of active orbitals
RECOMMENDATION:
The number of valence orbitals is a reasonable choice.
FON_T_SCALE
Determines the step size for the cooling.
TYPE:
INTEGER
DEFAULT:
90
OPTIONS:
n
temperature is scaled by
in each cycle (cooling method 1)
n
temperature is decreased by n K in each cycle (cooling method 2)
RECOMMENDATION:
The cooling rate should be neither too slow nor too fast. Too slow may lead to final energies that are at undesirably high temperatures. Too fast may lead to convergence issues. Reasonable choices for methods 1 and 2 are 98 and 50, respectively. When in doubt, use constant temperature.
FON_E_THRESH
DIIS error below which occupations will be kept constant.
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
n
freeze occupations below DIIS error of
RECOMMENDATION:
This should be one or two numbers bigger than the desired SCF convergence threshold.
FON_T_METHOD
Selects cooling algorithm.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
temperature is scaled by a factor in each cycle
2
temperature is decreased by a constant number in each cycle
RECOMMENDATION:
We have made slightly better experience with a constant cooling rate. However, choose constant temperature when in doubt.
4.5.11 Examples
Example 4.31 Input for a UHF calculation using geometric direct minimization (GDM) on the phenyl radical, after initial iterations with DIIS.
$molecule
0 2
c1
x1 c1 1.0
c2 c1 rc2 x1 90.0
x2 c2 1.0 c1 90.0 x1 0.0
c3 c1 rc3 x1 90.0 c2 tc3
c4 c1 rc3 x1 90.0 c2 -tc3
c5 c3 rc5 c1 ac5 x1 -90.0
c6 c4 rc5 c1 ac5 x1 90.0
h1 c2 rh1 x2 90.0 c1 180.0
h2 c3 rh2 c1 ah2 x1 90.0
h3 c4 rh2 c1 ah2 x1 -90.0
h4 c5 rh4 c3 ah4 c1 180.0
h5 c6 rh4 c4 ah4 c1 180.0
rc2 = 2.672986
rc3 = 1.354498
tc3 = 62.851505
rc5 = 1.372904
ac5 = 116.454370
rh1 = 1.085735
rh2 = 1.085342
ah2 = 122.157328
rh4 = 1.087216
ah4 = 119.523496
$end
$rem
BASIS = 6-31G*
METHOD = hf
SCF_ALGORITHM = diis_gdm
SCF_CONVERGENCE = 7
THRESH = 10
$end
Example 4.32 An example showing how to converge a ROHF calculation on the 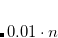 state of DMX. Note the use of reading in orbitals from a previous closed-shell calculation and the use of MOM to maintain the orbital occupancies. The
state of DMX. Note the use of reading in orbitals from a previous closed-shell calculation and the use of MOM to maintain the orbital occupancies. The 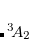 is obtained if MOM is not used.
is obtained if MOM is not used.
$molecule
+1 1
C 0.000000 0.000000 0.990770
H 0.000000 0.000000 2.081970
C -1.233954 0.000000 0.290926
C -2.444677 0.000000 1.001437
H -2.464545 0.000000 2.089088
H -3.400657 0.000000 0.486785
C -1.175344 0.000000 -1.151599
H -2.151707 0.000000 -1.649364
C 0.000000 0.000000 -1.928130
C 1.175344 0.000000 -1.151599
H 2.151707 0.000000 -1.649364
C 1.233954 0.000000 0.290926
C 2.444677 0.000000 1.001437
H 2.464545 0.000000 2.089088
H 3.400657 0.000000 0.486785
$end
$rem
UNRESTRICTED false
METHOD hf
BASIS 6-31+G*
SCF_GUESS core
$end
@@@
$molecule
read
$end
$rem
UNRESTRICTED false
METHOD hf
BASIS 6-31+G*
SCF_GUESS read
MOM_START 1
$end
$occupied
1:26 28
1:26 28
$end
@@@
$molecule
-1 3
read
$end
$rem
UNRESTRICTED false
METHOD hf
BASIS 6-31+G*
SCF_GUESS read
$end
Example 4.33 RCA_DIIS algorithm applied a radical
$molecule
0 2
H 1.004123 -0.180454 0.000000
O -0.246002 0.596152 0.000000
O -1.312366 -0.230256 0.000000
$end
$rem
UNRESTRICTED true
METHOD hf
BASIS cc-pVDZ
SCF_GUESS gwh
SCF_ALGORITHM RCA_DIIS
THRESH 9
$end
Example 4.34 pFON calculation of a metal cluster.
$molecule
0 1
Pt -0.20408 1.19210 0.54029
Pt 2.61132 1.04687 0.66196
Pt 0.83227 0.03296 -1.49084
Pt 0.95832 -1.05360 0.92253
Pt -1.66760 -1.07875 -1.02416
$end
$rem
METHOD pbe
ECP fit-lanl2dz
SYMMETRY false
OCCUPATIONS 2 ! pseudo-fractional occupation numbers
FON_NORB 10
FON_T_START 300 ! electronic temperature: 300 K
FON_T_END 300
FON_E_THRESH 5 ! freeze occupation numbers once DIIS error is 10^-5
$end