7.4 Maximum Overlap Method (MOM) for SCF Excited States
The Maximum Overlap Method (MOM) is a useful alternative to CIS and TDDFT for obtaining low-cost excited states.[Gilbert et al.(2008)Gilbert, Besley, and Gill] It works by modifying the orbital selection step in the SCF procedure. By choosing orbitals that most resemble those from the previous cycle, rather than those with the lowest eigenvalues, excited SCF determinants are able to be obtained. The MOM has several advantages over existing low-cost excited state methods. Current implementations of TDDFT usually struggle to accurately model charge-transfer and Rydberg transitions, both of which can be well-modeled using the MOM. The MOM also allows the user to target very high energy states, such as those involving excitation of core electrons,[Besley et al.(2009a)Besley, Gilbert, and Gill] which are hard to capture using other excited state methods.
In order to calculate an excited state using MOM, the user must correctly identify the orbitals involved in the transition. For example, in a 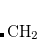 transition, the
transition, the 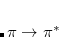 and
and  orbitals must be identified and this usually requires a preliminary calculation. The user then manipulates the orbital occupancies using the $occupied section, removing an electron from the and placing it in the . The MOM is then invoked to preserve this orbital occupancy. The success of the MOM relies on the quality of the initial guess for the calculation. If the virtual orbitals are of poor quality then the calculation may ‘fall down’ to a lower energy state of the same symmetry. Often the virtual orbitals of the corresponding cation are more appropriate for using as initial guess orbitals for the excited state.
orbitals must be identified and this usually requires a preliminary calculation. The user then manipulates the orbital occupancies using the $occupied section, removing an electron from the and placing it in the . The MOM is then invoked to preserve this orbital occupancy. The success of the MOM relies on the quality of the initial guess for the calculation. If the virtual orbitals are of poor quality then the calculation may ‘fall down’ to a lower energy state of the same symmetry. Often the virtual orbitals of the corresponding cation are more appropriate for using as initial guess orbitals for the excited state.
Because the MOM states are single determinants, all of Q-Chem’s existing single determinant properties and derivatives are available. This allows, for example, analytic harmonic frequencies to be computed on excited states. The orbitals from a Hartree-Fock MOM calculation can also be used in an MP2 calculation. For all excited state calculations, it is important to add diffuse functions to the basis set. This is particularly true if Rydberg transitions are being sought. For DFT based methods, it is also advisable to increase the size of the quadrature grid so that the more diffuse densities are accurately integrated.
Example 7.115 Calculation of the lowest singlet state of CO.
$comment
CO spin-purified calculation
$end
$molecule
0 1
C
O C 1.05
$end
$rem
METHOD B3LYP
BASIS 6-31G*
$end
@@@
$molecule
read
$end
$rem
METHOD B3LYP
BASIS 6-31G*
SCF_GUESS read
MOM_START 1
UNRESTRICTED true
OPSING true
$end
$occupied
1 2 3 4 5 6 7
1 2 3 4 5 6 8
$end
The following $rem is used to invoke the MOM:
MOM_START
Determines when MOM is switched on to preserve orbital occupancies.
TYPE:
INTEGER
DEFAULT:
0 (FALSE)
OPTIONS:
0 (FALSE)
MOM is not used

MOM begins on cycle
RECOMMENDATION:
For calculations on excited states, an initial calculation without MOM is usually required to get satisfactory starting orbitals. These orbitals should be read in using SCF_GUESS = true and MOM_START = 1.
Example 7.116 Input for obtaining the  A
A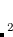 excited state of formamide corresponding to the transition. The
excited state of formamide corresponding to the transition. The  A ground state is obtained if MOM is not used in the second calculation. Note the use of diffuse functions and a larger quadrature grid to accurately model the larger excited state.
A ground state is obtained if MOM is not used in the second calculation. Note the use of diffuse functions and a larger quadrature grid to accurately model the larger excited state.
$molecule
1 2
C
H 1 1.091480
O 1 1.214713 2 123.10
N 1 1.359042 2 111.98 3 -180.00
H 4 0.996369 1 121.06 2 -0.00
H 4 0.998965 1 119.25 2 -180.00
$end
$rem
METHOD B3LYP
BASIS 6-311(2+,2+)G(d,p)
XC_GRID 000100000194
$end
@@@
$molecule
0 1
C
H 1 1.091480
O 1 1.214713 2 123.10
N 1 1.359042 2 111.98 3 -180.00
H 4 0.996369 1 121.06 2 -0.00
H 4 0.998965 1 119.25 2 -180.00
$end
$rem
METHOD B3LYP
BASIS 6-311(2+,2+)G(d,p)
XC_GRID 000100000194
MOM_START 1
SCF_GUESS read
UNRESTRICTED true
$end
$occupied
1:12
1:11 13
$end
Additionally, it is possible to perform a CIS/TDDFT calculation on top of the MOM excitation. This capability can be useful when modeling pump-probe spectra. In order to run MOM followed by CIS/TDDFT, the $rem variable cis_n_roots must be specified. Truncated subspaces may also be used, see Section 7.3.2.
Example 7.117 MOM valence excitation followed by core-state TDDFT using a restricted subspace
$molecule
0 1
O 0.0000 0.0000 0.1168
H 0.0000 0.7629 -0.4672
H 0.0000 -0.7629 -0.4672
$end
$rem
METHOD B3LYP
BASIS aug-cc-pvdz
SYMMETRY false
SYM_IGNORE true
$end
@@@
$molecule
read
$end
$rem
METHOD B3LYP
BASIS aug-cc-pvdz
SCF_GUESS read
MOM_START 1
UNRESTRICTED true
SYMMETRY false
SYM_IGNORE true
CIS_N_ROOTS 5
TRNSS true ! use truncated subspace for TDDFT
TRTYPE 3 ! specify occupied orbitals
CUTVIR 15 ! truncate high energy virtual orbitals
N_SOL 1 ! number core orbitals, specified in $solute section
$end
$solute
1
$end
$occupied
1 2 3 4 5
1 2 3 4 6
$end
If the MOM excitation corresponds to a core hole, a reduced subspace must be used to avoid de-excitations to the core hole. The $rem variable CORE_IONIZE allows only the hole to be specified so that not all occupied orbitals need to be entered in the $solute section.
CORE_IONIZE
Indicates how orbitals are specified for reduced excitation spaces.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
all valence orbitals are listed in $solute section
2
only hole(s) are specified all other occupations same as ground state
RECOMMENDATION:
For MOM + TDDFT this specifies the input form of the $solute section. If set to 1 all occupied orbitals must be specified, 2 only the empty orbitals to ignore must be specified.
Example 7.118 O(1s) core excited state using MOM followed by excitations among valence orbitals. Note that a reduced excitation subspace must be used to avoid “excitations” into the empty core hole
$molecule
0 1
O 0.0000 0.0000 0.1168
H 0.0000 0.7629 -0.4672
H 0.0000 -0.7629 -0.4672
$end
$rem
METHOD B3LYP
BASIS aug-cc-pvdz
SYMMETRY false
SYM_IGNORE true
$end
@@@
$molecule
read
$end
$rem
METHOD B3LYP
BASIS aug-cc-pvdz
SCF_GUESS read
MOM_START 1
UNRESTRICTED true
SYMMETRY false
SYMMETRY_IGNORE true
CIS_N_ROOTS 5
TRNSS true ! use truncated subspace for TDDFT
TRTYPE 3 ! specify occupied orbitals
N_SOL 1 ! number core holes, specified in $solute section
CORE_IONIZE 2 ! hole orbital specified
$end
$solute
6
$end
$occupied
1 2 3 4 5
2 3 4 5 6
$end