10.4 Potential Energy Scans
It is often useful to scan the potential energy surface (PES), optimizing all other degrees of freedom for each particular value of the scanned variable(s). Such a “relaxed” scan may provide a rough estimate of a pathway between reactant and product—assuming the coordinate(s) for the scan has been chosen wisely—and is often used in development of classical force fields to optimize dihedral angle parameters. Ramachandran plots, for example, are key tools for studying conformational changes of peptides and proteins, and are essentially two-dimensional torsional scans.
In certain cases, relaxed scans might encounter some difficulties on optimizations. A “frozen” scan can be easier to perform because of no geometry optimizations although it provides less information of real dynamics.
Q-Chem supports one- and two-dimensional PES scans, by setting JOBTYPE equal to PES_SCAN in the $rem section. In addition, a $scan input section with the following format should be specified, in the format below but with no more than two bond-length, bond-angle, or torsional variables specified.
$scan
stre atom1 atom2 value1 value2 incr
...
bend atom1 atom2 atom3 value1 value2 incr
...
tors atom1 atom2 atom3 atom4 value1 value2 incr
...
$end
The first example below demonstrates how to scan the torsional potential of butane, which is a sequence of constrained optimizations with the C1–C2–C3–C4 dihedral angle fixed at 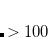 180
180 , 165, 150,
, 165, 150, 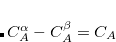 , 165, 180.
, 165, 180.
Example 10.217 One-dimensional torsional scan of butane
$molecule
0 1
C 1.934574 -0.128781 -0.000151
C 0.556601 0.526657 0.000200
C -0.556627 -0.526735 0.000173
C -1.934557 0.128837 -0.000138
H 2.720125 0.655980 -0.000236
H 2.061880 -0.759501 -0.905731
H 2.062283 -0.759765 0.905211
H 0.464285 1.168064 -0.903444
H 0.464481 1.167909 0.903924
H -0.464539 -1.167976 0.903964
H -0.464346 -1.168166 -0.903402
H -2.062154 0.759848 0.905185
H -2.720189 -0.655832 -0.000229
H -2.061778 0.759577 -0.905748
$end
$rem
JOBTYPE pes_scan
METHOD hf
BASIS sto-3g
$end
$scan
tors 1 2 3 4 -180 180 15
$end
The next example is a two-dimension potential scan. The first dimension is a scan of the C1–C2–C3–C4 dihedral angle from 180 to 180 degree in 30 intervals; the second dimension is a scan of the C2–C3 bond length from 1.5 Å to 1.6 Å in 0.05 Å increments.
Example 10.218 Two-dimensional torsional scan of butane
$molecule
0 1
C 1.934574 -0.128781 -0.000151
C 0.556601 0.526657 0.000200
C -0.556627 -0.526735 0.000173
C -1.934557 0.128837 -0.000138
H 2.720125 0.655980 -0.000236
H 2.061880 -0.759501 -0.905731
H 2.062283 -0.759765 0.905211
H 0.464285 1.168064 -0.903444
H 0.464481 1.167909 0.903924
H -0.464539 -1.167976 0.903964
H -0.464346 -1.168166 -0.903402
H -2.062154 0.759848 0.905185
H -2.720189 -0.655832 -0.000229
H -2.061778 0.759577 -0.905748
$end
$rem
JOBTYPE pes_scan
METHOD hf
BASIS sto-3g
$end
$scan
tors 1 2 3 4 -180 180 30
stre 2 3 1.5 1.6 0.05
$end
To perform a frozen PES scan, set FROZEN_SCAN to be TRUE and use input geometry in Z-matrix format. The example demonstrates a frozen PES of the C1–C2 bond stretching from 1.0 Åto 2.0 Åfor methanol.
Example 10.219 One-dimensional frozen PES scan of methanol
$molecule
0 1
C
O C RCO
H1 C RCH1 O H1CO
X C 1.0a O XCO H1 180.0
H2 C RCH2 X H2CX H1 90.0
H3 C RCH2 X H2CX H1 -90.0
H4 O ROH C HOC H1 180.0
RCO = 1.421
RCH1 = 1.094
RCH2 = 1.094
ROH = 0.963
H1CO = 107.2
XCO = 129.9
H2CX = 54.25
HOC = 108.0
$end
$rem
jobtype pes_scan
frozen_scan true
exchange s
correlatoin vwn
basis 3-21g
$end
$scan
stre 1 2 1.0 2.0 0.5
$end
Q-Chem also supports one-dimensional restrained PES scan for transition state search of typical S 2 reactions. The geometry restrains are
2 reactions. The geometry restrains are
 |
(10.1) |
which is a harmonic potential applied to bias geometry optimization.  and
and  are two bond lengths in the reaction coordinate.
are two bond lengths in the reaction coordinate. 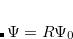 constrains the range of
constrains the range of  , and
, and 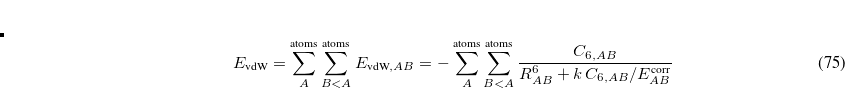 is a force constant. To perform a restrained PES scan, the following format should be specified.
is a force constant. To perform a restrained PES scan, the following format should be specified.
$scan
r12mr34 atom1 atom2 atom3 atom4 Rmin Rmax incr force_constant
r12pr34 atom1 atom2 atom3 atom4 Rmin Rmax incr force_constant
$end
Example 10.220 One-dimensional restrained PES scan of chloromethane SN2 reaction
$molecule
-1 1
C 0.418808 -1.240869 0.249048
Cl -0.775224 -1.495584 1.586668
H 1.408172 -1.490565 0.631227
H 0.147593 -1.907736 -0.568952
H 0.413296 -0.199000 -0.092071
Cl 1.947359 1.619163 -1.747832
$end
$rem
jobtype pes_scan
exchange b3lyp
basis 6-31G*
$end
$scan
r12mr34 1 2 1 6 -2.0 2.0 0.2 1000.0
$end