6.10 Coupled Cluster Active Space Methods
6.10.1 Introduction
Electron correlation effects can be qualitatively divided into two classes. The first class is static or non-dynamical correlation: long wavelength low-energy correlations associated with other electron configurations that are nearly as low in energy as the lowest energy configuration. These correlation effects are important for problems such as homolytic bond breaking, and are the hardest to describe because by definition the single configuration Hartree-Fock description is not a good starting point. The second class is dynamical correlation: short wavelength high-energy correlations associated with atomic-like effects. Dynamical correlation is essential for quantitative accuracy, but a reasonable description of static correlation is a prerequisite for a calculation being qualitatively correct.
In the methods discussed in the previous several subsections, the objective was to approximate the total correlation energy. However, in some cases, it is useful to model directly the non-dynamical and dynamical correlation energies separately. The reasons for this are pragmatic: with approximate methods, such a separation can give a more balanced treatment of electron correlation along bond-breaking coordinates, or reaction coordinates that involve diradicaloid intermediates. The non-dynamical correlation energy is conveniently defined as the solution of the Schrödinger equation within a small basis set composed of valence bonding, anti-bonding and lone pair orbitals: the so-called full valence active space. Solved exactly, this is the so-called full valence complete active space SCF (CASSCF),[Roos(1987)] or equivalently, the fully optimized reaction space (FORS) method.[Ruedenberg et al.(1982)Ruedenberg, Schmidt, Gilbert, and Elbert]
Full valence CASSCF and FORS involve computational complexity which increases exponentially with the number of atoms, and is thus unfeasible beyond systems of only a few atoms, unless the active space is further restricted on a case-by-case basis. Q-Chem includes two relatively economical methods that directly approximate these theories using a truncated coupled-cluster doubles wave function with optimized orbitals.[Krylov et al.(1998)Krylov, Sherrill, Byrd, and Head-Gordon] They are active space generalizations of the OD and QCCD methods discussed previously in Sections 6.8.3 and 6.8.4, and are discussed in the following two subsections. By contrast with the exponential growth of computational cost with problem size associated with exact solution of the full valence CASSCF problem, these cluster approximations have only 6th-order growth of computational cost with problem size, while often providing useful accuracy.
The full valence space is a well-defined theoretical chemical model. For these active space coupled-cluster doubles methods, it consists of the union of valence levels that are occupied in the single determinant reference, and those that are empty. The occupied levels that are to be replaced can only be the occupied valence and lone pair orbitals, whose number is defined by the sum of the valence electron counts for each atom (i.e., 1 for H, 2 for He, 1 for Li, etc..). At the same time, the empty virtual orbitals to which the double substitutions occur are restricted to be empty (usually anti-bonding) valence orbitals. Their number is the difference between the number of valence atomic orbitals, and the number of occupied valence orbitals given above. This definition (the full valence space) is the default when either of the “valence” active space methods are invoked (VOD or VQCCD)
There is also a second useful definition of a valence active space, which we shall call the 1:1 or perfect pairing active space. In this definition, the number of occupied valence orbitals remains the same as above. The number of empty correlating orbitals in the active space is defined as being exactly the same number, so that each occupied orbital may be regarded as being associated 1:1 with a correlating virtual orbital. In the water molecule, for example, this means that the lone pair electrons as well as the bond-orbitals are correlated. Generally the 1:1 active space recovers more correlation for molecules dominated by elements on the right of the periodic table, while the full valence active space recovers more correlation for molecules dominated by atoms to the left of the periodic table.
If you wish to specify either the 1:1 active space as described above, or some other choice of active space based on your particular chemical problem, then you must specify the numbers of active occupied and virtual orbitals. This is done via the standard “window options”, documented earlier in this Chapter.
Finally we note that the entire discussion of active spaces here leads only to specific numbers of active occupied and virtual orbitals. The orbitals that are contained within these spaces are optimized by minimizing the trial energy with respect to all the degrees of freedom previously discussed: the substitution amplitudes, and the orbital rotation angles mixing occupied and virtual levels. In addition, there are new orbital degrees of freedom to be optimized to obtain the best active space of the chosen size, in the sense of yielding the lowest coupled-cluster energy. Thus rotation angles mixing active and inactive occupied orbitals must be varied until the energy is stationary. Denoting inactive orbitals by primes and active orbitals without primes, this corresponds to satisfying
 |
(6.43) |
Likewise, the rotation angles mixing active and inactive virtual orbitals must also be varied until the coupled-cluster energy is minimized with respect to these degrees of freedom:
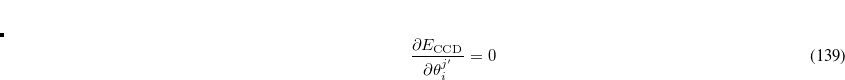 |
(6.44) |
6.10.2 VOD and VOD(2) Methods
The VOD method is the active space version of the OD method described earlier in Section 6.8.3. Both energies and gradients are available for VOD, so structure optimization is possible. There are a few important comments to make about the usefulness of VOD. First, it is a method that is capable of accurately treating problems that fundamentally involve 2 active electrons in a given local region of the molecule. It is therefore a good alternative for describing single bond-breaking, or torsion around a double bond, or some classes of diradicals. However it often performs poorly for problems where there is more than one bond being broken in a local region, with the non variational solutions being quite possible. For such problems the newer VQCCD method is substantially more reliable.
Assuming that VOD is a valid zero order description for the electronic structure, then a second-order correction, VOD(2), is available for energies only. VOD(2) is a version of OD(2) generalized to valence active spaces. It permits more accurate calculations of relative energies by accounting for dynamical correlation.
6.10.3 VQCCD
The VQCCD method is the active space version of the QCCD method described earlier in Section 6.8.3. Both energies and gradients are available for VQCCD, so that structure optimization is possible. VQCCD is applicable to a substantially wider range of problems than the VOD method, because the modified energy functional is not vulnerable to non variational collapse. Testing to date suggests that it is capable of describing double bond breaking to similar accuracy as full valence CASSCF, and that potential curves for triple bond-breaking are qualitatively correct, although quantitatively in error by a few tens of kcal/mol. The computational cost scales in the same manner with system size as the VOD method, albeit with a significantly larger prefactor.
6.10.4 CCVB-SD
Working with Prof. Head-Gordon at Berkeley, Dr. D. W. Small and Joonho Lee have developed and implemented a novel single-reference coupled-cluster method with singles and doubles, called CCVB-SD.[Small and Head-Gordon(2012), J. Lee and Head-Gordon(2017)] CCVB-SD improves upon a more crude model CCVB (Section 6.15.2) and can be considered a simple modification to restricted CCSD (RCCSD). CCVB-SD inherits good properties from CCVB and RCCSD; it is spin-pure, size-extensive, and capable of breaking multiple bonds as long as only the valence space is correlated. It is a full doubles model and thus scales 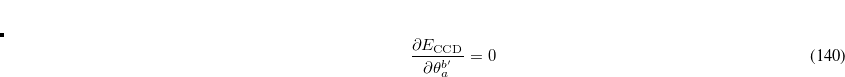 . However, its energy is invariant under rotations in occupied space and virtual space, which makes it much more black-box than CCVB. Its energy function follows
. However, its energy is invariant under rotations in occupied space and virtual space, which makes it much more black-box than CCVB. Its energy function follows
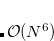 |
(6.45) |
where 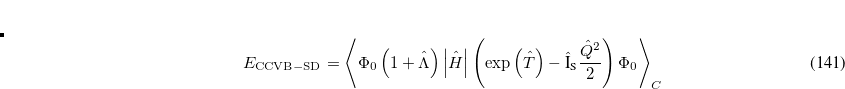 is a singlet projection operator and
is a singlet projection operator and 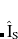 is a quintet doubles operator. Unlike QCCD, CCVB-SD improves the right eigenfunction while leaving the left eigenfunction unchanged. The quintet term in Eq. eq:ccvbsd represents approximate connected quadruples which are responsible for describing strong correlation. The cost of CCVB-SD is only twice as expensive as RCCSD, and it is better suited for strong correlation than QCCD/VQCCD in the sense that the method becomes exact at the dissociation limits of most multiple bond breaking whereas QCCD does not except special cases.
is a quintet doubles operator. Unlike QCCD, CCVB-SD improves the right eigenfunction while leaving the left eigenfunction unchanged. The quintet term in Eq. eq:ccvbsd represents approximate connected quadruples which are responsible for describing strong correlation. The cost of CCVB-SD is only twice as expensive as RCCSD, and it is better suited for strong correlation than QCCD/VQCCD in the sense that the method becomes exact at the dissociation limits of most multiple bond breaking whereas QCCD does not except special cases.
Although CCVB-SD can be used without the active space constraints, we recommend that users use it with the valence active space in general. For benchmarking purposes, using a minimal basis will automatically provide the valence space correctly with frozen cores. Both the energy and nuclear gradients of CCVB-SD are available through CCMAN2.
It should be noted that there is no orbital optimization implemented for CCVB-SD at the moment. This means that using basis sets larger than minimal basis requires choosing right valence orbitals to use. Therefore, we recommend that users run GVB-PP (or CCVB) to obtain orbitals to begin with. Orbital optimization (i.e. CCVB-OD) will soon be implemented and running CCVB-OD will be much more black-box than CCVB-SD as it does not require selecting proper valence space orbitals.
Furthermore, CCVB-SD can be applied to only closed-shell molecules at the moment. The extension to open-shell molecules is under development.
Example 6.91 A CCVB-SD force calculation of benzene in a minimal basis.
$comment
CCVB-SD job for benzene
It will compute energy+gradients.
It will also print out natural orbital occupation numbers (NOONs)
$end
$molecule
0 1
C 0.000000 0.698200 0.000000
C 0.000000 -0.698200 0.000000
C 1.209318 1.396400 0.000000
C 1.209318 -1.396400 0.000000
C 2.418636 0.698200 0.000000
C 2.418636 -0.698200 0.000000
H -0.931410 1.235950 0.000000
H -0.931410 -1.235950 0.000000
H 1.209318 2.471900 0.000000
H 1.209318 -2.471900 0.000000
H 3.350046 1.235950 0.000000
H 3.350046 -1.235950 0.000000
$end
$rem
JOBTYPE = force
BASIS = sto-3g
METHOD = ccvbsd
THRESH = 14
SCF_ALGORITHM = gdm
SCF_CONVERGENCE = 10
CC_REF_PROP = true
SYMMETRY = false
SYM_IGNORE = true
$end
6.10.5 Local Pair Models for Valence Correlations Beyond Doubles
Working with Prof. Head-Gordon at Berkeley, John Parkhill has developed implementations for pair models which couple 4 and 6 electrons together quantitatively. Because these truncate the coupled cluster equations at quadruples and hextuples respectively they have been termed the “Perfect Quadruples” and “Perfect Hextuples” models. These can be viewed as local approximations to CASSCF. The PQ and PH models are executed through an extension of Q-Chem’s coupled cluster code, and several options defined for those models will have the same effects although the mechanism may be different (CC_DIIS_START, CC_DIIS_SIZE, CC_DOV_THRESH, CC_CONV, etc.).
In the course of implementation, the non-local coupled cluster models were also implemented up to 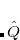 . Because the algorithms are explicitly sparse their costs relative to the existing implementations of CCSD are much higher (and should never be used in lieu of an existing CCMAN code), but this capability may be useful for development purposes, and when computable, models above CCSDTQ are highly accurate. To use PQ, PH, their dynamically correlated “+SD” versions or this machine generated cluster code set: METHOD = MGC.
. Because the algorithms are explicitly sparse their costs relative to the existing implementations of CCSD are much higher (and should never be used in lieu of an existing CCMAN code), but this capability may be useful for development purposes, and when computable, models above CCSDTQ are highly accurate. To use PQ, PH, their dynamically correlated “+SD” versions or this machine generated cluster code set: METHOD = MGC.
MGC_AMODEL
Choice of approximate cluster model.
TYPE:
INTEGER
DEFAULT:
Determines
how the CC equations are approximated:
OPTIONS:
0
Local Active-Space Amplitude iterations (pre-calculate GVB orbitals with your method of choice (RPP is good)).
7
Optimize-Orbitals using the VOD 2-step solver.
(Experimental-only use with MGC_AMPS = 2, 24 ,246)
8
Traditional Coupled Cluster up to CCSDTQPH.
9
MR-CC version of the Pair-Models. (Experimental)
RECOMMENDATION:
None
MGC_NLPAIRS
Number of local pairs on an amplitude.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
Must be greater than 1, which corresponds to the PP model. 2 for PQ, and 3 for PH.
RECOMMENDATION:
None
MGC_AMPS
Choice of Amplitude Truncation
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
2
n
which will be iterated. Choose 1234 for PQ and 123456 for PH
RECOMMENDATION:
None
MGC_LOCALINTS
Pair filter on an integrals.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Enforces a pair filter on the 2-electron integrals, significantly
reducing computational cost. Generally useful. for more than 1 pair locality.
RECOMMENDATION:
None
MGC_LOCALINTER
Pair filter on an intermediate.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Any nonzero value enforces the pair constraint on intermediates,
significantly reducing computational cost. Not recommended for
RECOMMENDATION:
None
6.10.6 Convergence Strategies and More Advanced Options
These optimized orbital coupled-cluster active space methods enable the use of the full valence space for larger systems than is possible with conventional complete active space codes. However, we should note at the outset that often there are substantial challenges in converging valence active space calculations (and even sometimes optimized orbital coupled cluster calculations without an active space). Active space calculations cannot be regarded as “routine” calculations in the same way as SCF calculations, and often require a considerable amount of computational trial and error to persuade them to converge. These difficulties are largely because of strong coupling between the orbital degrees of freedom and the amplitude degrees of freedom, as well as the fact that the energy surface is often quite flat with respect to the orbital variations defining the active space.
Being aware of this at the outset, and realizing that the program has nothing against you personally is useful information for the uninitiated user of these methods. What the program does have, to assist in the struggle to achieve a converged solution, are accordingly many convergence options, fully documented in Appendix C. In this section, we describe the basic options and the ideas behind using them as a starting point. Experience plays a critical role, however, and so we encourage you to experiment with toy jobs that give rapid feedback in order to become proficient at diagnosing problems.
If the default procedure fails to converge, the first useful option to employ is CC_PRECONV_T2Z, with a value of between 10 and 50. This is useful for jobs in which the MP2 amplitudes are very poor guesses for the converged cluster amplitudes, and therefore initial iterations varying only the amplitudes will be beneficial:
CC_PRECONV_T2Z
Whether to pre-converge the cluster amplitudes before beginning orbital optimization in optimized orbital cluster methods.
TYPE:
INTEGER
DEFAULT:
0
(FALSE)
10
If CC_RESTART, CC_RESTART_NO_SCF or CC_MP2NO_GUESS are TRUE
OPTIONS:
0
No pre-convergence before orbital optimization.
Up to
iterations in this pre-convergence procedure.
RECOMMENDATION:
Experiment with this option in cases of convergence failure.
Other options that are useful include those that permit some damping of step sizes, and modify or disable the standard DIIS procedure. The main choices are as follows.
CC_DIIS
Specify the version of Pulay’s Direct Inversion of the Iterative Subspace (DIIS) convergence accelerator to be used in the coupled-cluster code.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Activates procedure 2 initially, and procedure 1 when gradients are smaller
than DIIS12_SWITCH.
1
Uses error vectors defined as differences between parameter vectors from
successive iterations. Most efficient near convergence.
2
Error vectors are defined as gradients scaled by square root of the
approximate diagonal Hessian. Most efficient far from convergence.
RECOMMENDATION:
DIIS1 can be more stable. If DIIS problems are encountered in the early stages of a calculation (when gradients are large) try DIIS1.
CC_DIIS_START
Iteration number when DIIS is turned on. Set to a large number to disable DIIS.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
User-defined
RECOMMENDATION:
Occasionally DIIS can cause optimized orbital coupled-cluster calculations to diverge through large orbital changes. If this is seen, DIIS should be disabled.
CC_DOV_THRESH
Specifies minimum allowed values for the coupled-cluster energy denominators. Smaller values are replaced by this constant during early iterations only, so the final results are unaffected, but initial convergence is improved when the guess is poor.
TYPE:
INTEGER
DEFAULT:
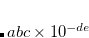
Corresponding to 0.25
OPTIONS:
Integer code is mapped to
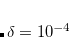
RECOMMENDATION:
Increase to 0.5 or 0.75 for non convergent coupled-cluster calculations.
CC_THETA_STEPSIZE
Scale factor for the orbital rotation step size. The optimal rotation steps should be approximately equal to the gradient vector.
TYPE:
INTEGER
DEFAULT:
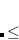
Corresponding to 1.0
OPTIONS:
Integer code is mapped to
If the initial step is smaller than 0.5, the program will increase step
when gradients are smaller than the value of THETA_GRAD_THRESH,
up to a limit of 0.5.
RECOMMENDATION:
Try a smaller value in cases of poor convergence and very large orbital gradients. For example, a value of 01001 translates to 0.1
An even stronger—and more-or-less last resort—option permits iteration of the cluster amplitudes without changing the orbitals:
CC_PRECONV_T2Z_EACH
Whether to pre-converge the cluster amplitudes before each change of the orbitals in optimized orbital coupled-cluster methods. The maximum number of iterations in this pre-convergence procedure is given by the value of this parameter.
TYPE:
INTEGER
DEFAULT:
0
(FALSE)
OPTIONS:
0
No pre-convergence before orbital optimization.
Up to
RECOMMENDATION:
A very slow last resort option for jobs that do not converge.
6.10.7 Examples
Example 6.92 Two jobs that compare the correlation energy of the water molecule with partially stretched bonds, calculated via the two coupled-cluster active space methods, VOD, and VQCCD. These are relatively “easy” jobs to converge, and may be contrasted with the next example, which is not easy to converge. The orbitals are restricted.
$molecule
0 1
O
H 1 r
H 1 r a
r = 1.5
a = 104.5
$end
$rem
METHOD vod
BASIS 6-31G
$end
@@@
$molecule
read
$end
$rem
METHOD vqccd
BASIS 6-31G
$end
Example 6.93 The water molecule with highly stretched bonds, calculated via the two coupled-cluster active space methods, VOD, and VQCCD. These are “difficult” jobs to converge. The convergence options shown permitted the job to converge after some experimentation (thanks due to Ed Byrd for this!). The difficulty of converging this job should be contrasted with the previous example where the bonds were less stretched. In this case, the VQCCD method yields far better results than VOD!.
$molecule
0 1
O
H 1 r
H 1 r a
r = 3.0
a = 104.5
$end
$rem
METHOD vod
BASIS 6-31G
SCF_CONVERGENCE 9
THRESH 12
CC_PRECONV_T2Z 50
CC_PRECONV_T2Z_EACH 50
CC_DOV_THRESH 7500
CC_THETA_STEPSIZE 3200
CC_DIIS_START 75
$end
@@@
$molecule
read
$end
$rem
METHOD vqccd
BASIS 6-31G
SCF_CONVERGENCE 9
THRESH 12
CC_PRECONV_T2Z 50
CC_PRECONV_T2Z_EACH 50
CC_DOV_THRESH 7500
CC_THETA_STEPSIZE 3200
CC_DIIS_START 75
$end