13.16 TDDFT for Molecular Interactions
13.16.1 Theory
There exist a broad class of weakly interacting molecular complexes which give rise to interesting excited-state properties that are potentially very different from those of a single chromophore. The “TDDFT for molecular interactions" or TDDFT(MI) method is designed for efficient excited-state calculations in such cases in (potentially large) systems composed of weakly-interacting but electronically-coupled monomers.[Liu and Herbert(2015), Herbert et al.(2016)Herbert, Zhang, Morrison, and Liu] Such systems include molecular aggregates, chromophores in explicit solvent, and even proteins, for which the traditional TDDFT method become prohibitively expensive. TDDFT(MI) starts from a ground-state SCF MI calculation, and the use of ALMOs is central to its efficiency. In addition, the excitations are confined within monomer units and the explicit charge-transfer excitations are ignored, significantly reduced the two-electron integrals cost. The method works by coupling together excitations computed individually on different molecular fragments, and the number of excited states per fragment can be increased (at very low cost) in order to increase the variational flexibility of this exciton-type basis. Thus, despite the localized nature of the basis states, TDDFT(MI) is capable of describing collective excitations that are delocalized over multiple monomer units, as for example in the case of organic semiconductors. In general, TDDFT(MI) reproduces full super-system TDDFT excitation energies to within 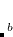 0.2 eV, but with an order or magnitude reduction in total CPU time.[Liu and Herbert(2015)] Formally, the cost of the method scales as
0.2 eV, but with an order or magnitude reduction in total CPU time.[Liu and Herbert(2015)] Formally, the cost of the method scales as  where
where  is the number of monomers,
is the number of monomers,  is the number of excited states per monomer, and
is the number of excited states per monomer, and  is the number of AOs on a dimer subsystem. The exponent
is the number of AOs on a dimer subsystem. The exponent 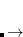 (with
(with  ) reflects the cost of forming the Fock-like matrices of a traditional TDDFT calculation.
) reflects the cost of forming the Fock-like matrices of a traditional TDDFT calculation.
An especially promising application of the TDDFT(MI) method is to study excitation energies of a single chromophore in solution using a large number of explicit, quantum-mechanical solvent molecules. In such cases, the excitations are localized on the single chromophore and we can introduce a local excitation approximation (LEA) to TDDFT(MI) in which all of the Coulomb and exchange couplings between the solvent molecules and the chromophore are neglected.[Liu and Herbert(2016)] Following the ground-state SCF(MI) calculation, the cost of the TDDFT part of the calculation becomes essentially the same as the cost of a TDDFT calculation on the gas-phase chromophore. In addition, this approach avoids the appearance of, and mixing with, spurious charge-transfer-to-solvent states,[Liu and Herbert(2016), Herbert et al.(2016)Herbert, Zhang, Morrison, and Liu] of the sort that are known to arise in TDDFT calculations with explicit solvent.[Lange and Herbert(2007), Isborn et al.(2013)Isborn, Mar, Curchod, Tavernelli, and Mart nez] Three versions of LEA-TDDFT(MI), named LEA0, LEA-Q and LEAc, have been implemented in Q-Chem.[Liu and Herbert(2016)] In the LEA0 method, ALMOs from the ground state SCF(MI) calculation are used to perform the TDDFT calculation. In LEAc, a sub-block of the TDDFT(MI) working equation localized on chromophore is extracted to calculate the excitation energies. Finally, LEA-Q is almost the same as LEAc except for some transformations to eliminate the overlap matrices. These approaches have been applied to converge solvatochromatic shifts for several aqueous chromophores.[Liu and Herbert(2016)]
nez] Three versions of LEA-TDDFT(MI), named LEA0, LEA-Q and LEAc, have been implemented in Q-Chem.[Liu and Herbert(2016)] In the LEA0 method, ALMOs from the ground state SCF(MI) calculation are used to perform the TDDFT calculation. In LEAc, a sub-block of the TDDFT(MI) working equation localized on chromophore is extracted to calculate the excitation energies. Finally, LEA-Q is almost the same as LEAc except for some transformations to eliminate the overlap matrices. These approaches have been applied to converge solvatochromatic shifts for several aqueous chromophores.[Liu and Herbert(2016)]
13.16.2 Job Control
In addition to the normal TDDFT job controls variables described in Section 7.3.3, there are several others to request TDDFT(MI). Note that only single-point energies (not gradients) are available for this method.
TDDFT_MI
Perform an TDDFT(MI) calculation
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform an TDDFT(MI) calculation
TRUE
Perform an TDDFT(MI) calculation
RECOMMENDATION:
False
MI_ACTIVE_FRAGMENT
Sets the active fragment
TYPE:
INTEGER
DEFAULT:
NO DEFAULT
OPTIONS:

Specify the fragment on which the TDDFT calculation is to be performed, for LEA-TDDFT(MI).
RECOMMENDATION:
None
MI_LEA
Controls the LEA-TDDFT(MI) methods
TYPE:
INTEGER
DEFAULT:
NO DEFAULT
OPTIONS:
0
The LEA0 method
1
The LEA-Q method
2
The LEAc method
RECOMMENDATION:
1