C.4 Alphabetical Listing of $rem Variables
EIGSLV_METH
Control the method for solving the ALMO-CIS eigen-equation
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Explicitly build the Hamiltonian then diagonalize (full-spectrum).
1
Use the Davidson method (currently only available for restricted cases).
RECOMMENDATION:
None
LOCAL_CIS
Invoke ALMO-CIS/ALMO-CIS+CT.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Regular CIS
1
ALMO-CIS/ALMO-CIS+CT without RI(slow)
2
ALMO-CIS/ALMO-CIS+CT with RI
RECOMMENDATION:
2 if ALMO-CIS is desired.
NN_THRESH
The distance cutoff for neighboring fragments (between which CT is enabled).
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not include interfragment transitions (ALMO-CIS).

Include interfragment excitations between pairs of fragments the distances between whom
are smaller than
RECOMMENDATION:
None
FDIFF_STEPSIZE
Displacement used for calculating derivatives by finite difference.
TYPE:
INTEGER
DEFAULT:
1
Corresponding to
a.u.
OPTIONS:
Use a step size of
RECOMMENDATION:
Use the default unless problems arise.
RESPONSE_POLAR
Control the use of analytic or numerical polarizabilities.
TYPE:
INTEGER
DEFAULT:
0 or
1
= 0 for HF or DFT,
OPTIONS:
0
Perform an analytic polarizability calculation.
Perform a numeric polarizability calculation even when analytic 2nd derivatives are available.
RECOMMENDATION:
None
ADC_CVS
Activates the use of the CVS approximation for the calculation of CVS-ADC core-excited states.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Activates the CVS approximation.
FALSE
Do not compute core-excited states using the CVS approximation.
RECOMMENDATION:
Set to TRUE, if to obtain core-excited states for the simulation of X-ray absorption spectra. In the case of TRUE, the $rem variable CC_REST_OCC has to be defined as well.
ADC_C_C
Set the spin-opposite scaling parameter
for the ADC(2) calculation. The parameter value is obtained by multiplying the given integer by
.
TYPE:
INTEGER
DEFAULT:
1170
Optimized value
for ADC(2)-s or
1000
for ADC(2)-x
OPTIONS:
Corresponding to

RECOMMENDATION:
Use the default.
ADC_C_T
Set the spin-opposite scaling parameter
for an SOS-ADC(2) calculation. The parameter value is obtained by multiplying the given integer by
TYPE:
INTEGER
DEFAULT:
1300
Optimized value
.
OPTIONS:
Corresponding to
RECOMMENDATION:
Use the default.
ADC_C_X
Set the spin-opposite scaling parameter
for the ADC(2)-x calculation. The parameter value is obtained by multiplying the given integer by
TYPE:
INTEGER
DEFAULT:
1300
Optimized value
for ADC(2)-x.
OPTIONS:
Corresponding to
RECOMMENDATION:
Use the default.
ADC_DAVIDSON_CONV
Controls the convergence criterion of the Davidson procedure.
TYPE:
INTEGER
DEFAULT:

Corresponding to
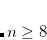
OPTIONS:

Corresponding to
.
RECOMMENDATION:
Use the default unless higher accuracy is required or convergence problems are encountered.
ADC_DAVIDSON_MAXITER
Controls the maximum number of iterations of the Davidson procedure.
TYPE:
INTEGER
DEFAULT:
60
OPTIONS:
Number of iterations
RECOMMENDATION:
Use the default unless convergence problems are encountered.
ADC_DAVIDSON_MAXSUBSPACE
Controls the maximum subspace size for the Davidson procedure.
TYPE:
INTEGER
DEFAULT:
the number of excited states to be calculated.
OPTIONS:
User-defined integer.
RECOMMENDATION:
Should be at least
the number of excited states to calculate. The larger the value the more disk space is required.
ADC_DAVIDSON_THRESH
Controls the threshold for the norm of expansion vectors to be added during the Davidson procedure.
TYPE:
INTEGER
DEFAULT:
Twice the value of ADC_DAVIDSON_CONV, but at maximum
.
OPTIONS:

Corresponding to
RECOMMENDATION:
Use the default unless convergence problems are encountered. The threshold value
ADC_DIIS_ECONV
Controls the convergence criterion for the excited state energy during DIIS.
TYPE:
INTEGER
DEFAULT:
6
Corresponding to
OPTIONS:
Corresponding to
RECOMMENDATION:
None
ADC_DIIS_MAXITER
Controls the maximum number of DIIS iterations.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
User-defined integer.
RECOMMENDATION:
Increase in case of slow convergence.
ADC_DIIS_RCONV
Convergence criterion for the residual vector norm of the excited state during DIIS.
TYPE:
INTEGER
DEFAULT:
6
Corresponding to
OPTIONS:
Corresponding to
RECOMMENDATION:
None
ADC_DIIS_SIZE
Controls the size of the DIIS subspace.
TYPE:
INTEGER
DEFAULT:
7
OPTIONS:
User-defined integer
RECOMMENDATION:
None
ADC_DIIS_START
Controls the iteration step at which DIIS is turned on.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
User-defined integer.
RECOMMENDATION:
Set to a large number to switch off DIIS steps.
ADC_DO_DIIS
Activates the use of the DIIS algorithm for the calculation of ADC(2) excited states.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Use DIIS algorithm.
FALSE
Do diagonalization using Davidson algorithm.
RECOMMENDATION:
None.
ADC_NGUESS_DOUBLES
Controls the number of excited state guess vectors which are double excitations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined integer.
RECOMMENDATION:
ADC_NGUESS_SINGLES
Controls the number of excited state guess vectors which are single excitations. If the number of requested excited states exceeds the total number of guess vectors (singles and doubles), this parameter is automatically adjusted, so that the number of guess vectors matches the number of requested excited states.
TYPE:
INTEGER
DEFAULT:
Equals to the number of excited states requested.
OPTIONS:
User-defined integer.
RECOMMENDATION:
ADC_PRINT
Controls the amount of printing during an ADC calculation.
TYPE:
INTEGER
DEFAULT:
1
Basic status information and results are printed.
OPTIONS:
0
Quiet: almost only results are printed.
1
Normal: basic status information and results are printed.
2
Debug: more status information, extended information on timings.
...
RECOMMENDATION:
Use the default.
ADC_PROP_ES2ES
Controls the calculation of transition properties between excited states (currently only transition dipole moments and oscillator strengths), as well as the computation of two-photon absorption cross-sections of excited states using the sum-over-states expression.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculate state-to-state transition properties.
FALSE
Do not compute transition properties between excited states.
RECOMMENDATION:
Set to TRUE, if state-to-state properties or sum-over-states two-photon absorption cross-sections are required.
ADC_PROP_ES
Controls the calculation of excited state properties (currently only dipole moments).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculate excited state properties.
FALSE
Do not compute state properties.
RECOMMENDATION:
Set to TRUE, if properties are required.
ADC_PROP_TPA
Controls the calculation of two-photon absorption cross-sections of excited states using matrix inversion techniques.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculate two-photon absorption cross-sections.
FALSE
Do not compute two-photon absorption cross-sections.
RECOMMENDATION:
Set to TRUE, if to obtain two-photon absorption cross-sections.
ADD_CHARGED_CAGE
Add a point charge cage of a given radius and total charge.
TYPE:
INTEGER
DEFAULT:
0
No cage.
OPTIONS:
0
No cage.
1
Dodecahedral cage.
2
Spherical cage.
RECOMMENDATION:
Spherical cage is expected to yield more accurate results, especially for small radii.
AFSSH
Adds decoherence approximation to surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Traditional surface hopping, no decoherence.
1
Use augmented fewest-switches surface hopping (AFSSH).
RECOMMENDATION:
AFSSH will increase the cost of the calculation, but may improve accuracy for some systems. See Refs. Subotnik:2011a,Subotnik:2011b,Landry:2012 for more detail.
AIFDEM_CTSTATES
Include charge-transfer-like cation/anion pair states in the AIFDEM basis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Include CT states.
FALSE
Do not include CT states.
RECOMMENDATION:
None
AIFDEM_EMBED_RANGE
Specifies the size of the QM region for charge embedding
TYPE:
INTEGER
DEFAULT:
FULL_QM
OPTIONS:
FULL_QM
No charge embedding.
0
Treat only excited fragments with QM.
Range (Å) from excited fragments within which to treat other fragments with QM.
RECOMMENDATION:
Minimal, 0 Å, threshold maintains accuracy while significantly reducing computational time.
AIFDEM_NTOTHRESH
Controls the number of NTOs that are retained in the exciton-site basis states.
TYPE:
INTEGER
DEFAULT:
99
OPTIONS:
Threshold percentage of the norm of fragment NTO amplitudes.
RECOMMENDATION:
A threshold of
gives a good trade-off of computational time and accuracy for organic molecules.
AIFDEM
Perform an AIFDEM calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform an AIFDEM calculation.
TRUE
Perform an AIFDEM calculation.
RECOMMENDATION:
False
AIMD_FICT_MASS
Specifies the value of the fictitious electronic mass
, in atomic units, where
(time)
.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
User-specified
RECOMMENDATION:
Values in the range of 50–200 a.u. have been employed in test calculations; consult Ref. Herbert:2004 for examples and discussion.
AIMD_INIT_VELOC
Specifies the method for selecting initial nuclear velocities.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
THERMAL
Random sampling of nuclear velocities from a Maxwell-Boltzmann
distribution. The user must specify the temperature in Kelvin via
the $rem variable AIMD_TEMP.
ZPE
Choose velocities in order to put zero-point vibrational energy into
each normal mode, with random signs. This option requires that a
frequency job to be run beforehand.
QUASICLASSICAL
Puts vibrational energy into each normal mode. In contrast to the
ZPE option, here the vibrational energies are sampled from a
Boltzmann distribution at the desired simulation temperature. This
also triggers several other options, as described below.
RECOMMENDATION:
This variable need only be specified in the event that velocities are not specified explicitly in a $velocity section.
AIMD_LANGEVIN_TIMESCALE
Sets the timescale (strength) of the Langevin thermostat
TYPE:
INTEGER
DEFAULT:
none
OPTIONS:
Thermostat timescale,asn
RECOMMENDATION:
Smaller values (roughly 100) equate to tighter thermostats but may inhibit rapid sampling. Larger values (
) allow for more rapid sampling but may take longer to reach thermal equilibrium.
AIMD_METHOD
Selects an ab initio molecular dynamics algorithm.
TYPE:
STRING
DEFAULT:
BOMD
OPTIONS:
BOMD
Born-Oppenheimer molecular dynamics.
CURVY
Curvy-steps Extended Lagrangian molecular dynamics.
RECOMMENDATION:
BOMD yields exact classical molecular dynamics, provided that the energy is tolerably conserved. ELMD is an approximation to exact classical dynamics whose validity should be tested for the properties of interest.
AIMD_MOMENTS
Requests that multipole moments be output at each time step.
TYPE:
INTEGER
DEFAULT:
0
Do not output multipole moments.
OPTIONS:
Output the first
RECOMMENDATION:
None
AIMD_NUCL_DACF_POINTS
Number of time points to use in the dipole auto-correlation function for an AIMD trajectory
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not compute dipole auto-correlation function.

Compute dipole auto-correlation function for last
timesteps of the trajectory.
RECOMMENDATION:
If the DACF is desired, set equal to AIMD_STEPS.
AIMD_NUCL_SAMPLE_RATE
The rate at which sampling is performed for the velocity and/or dipole auto-correlation function(s). Specified as a multiple of steps; i.e., sampling every step is 1.
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
Update the velocity/dipole auto-correlation function
every
RECOMMENDATION:
Since the velocity and dipole moment are routinely calculated for ab initio methods, this variable should almost always be set to 1 when the VACF/DACF are desired.
AIMD_NUCL_VACF_POINTS
Number of time points to use in the velocity auto-correlation function for an AIMD trajectory
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not compute velocity auto-correlation function.
Compute velocity auto-correlation function for last
time steps of the trajectory.
RECOMMENDATION:
If the VACF is desired, set equal to AIMD_STEPS.
AIMD_QCT_INITPOS
Chooses the initial geometry in a QCT-MD simulation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
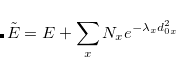
Use the equilibrium geometry.
Picks a random geometry according to the harmonic vibrational wave function.
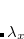
Generates
the harmonic vibrational wave function.
RECOMMENDATION:
None.
AIMD_QCT_WHICH_TRAJECTORY
Picks a set of vibrational quantum numbers from a random distribution.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
Picks the
Uses an average over
RECOMMENDATION:
Pick a positive number if you want the initial velocities to correspond to a particular set of vibrational occupation numbers and choose a different number for each of your trajectories. If initial velocities are desired that corresponds to an average over
AIMD_SHORT_TIME_STEP
Specifies a shorter electronic time step for FSSH calculations.
TYPE:
INTEGER
DEFAULT:
TIME_STEP
OPTIONS:
Specify an electronic time step duration of
a.u. If
electronic wave function will be integrated multiple times per nuclear time step,
using a linear interpolation of nuclear quantities such as the energy gradient and
derivative coupling. Note that
RECOMMENDATION:
Make AIMD_SHORT_TIME_STEP as large as possible while keeping the trace of the density matrix close to unity during long simulations. Note that while specifying an appropriate duration for the electronic time step is essential for maintaining accurate wave function time evolution, the electronic-only time steps employ linear interpolation to estimate important quantities. Consequently, a short electronic time step is not a substitute for a reasonable nuclear time step.
AIMD_STEPS
Specifies the requested number of molecular dynamics steps.
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
User-specified.
RECOMMENDATION:
None.
AIMD_TEMP
Specifies a temperature (in Kelvin) for Maxwell-Boltzmann velocity sampling.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
User-specified number of Kelvin.
RECOMMENDATION:
This variable is only useful in conjunction with AIMD_INIT_VELOC = THERMAL. Note that the simulations are run at constant energy, rather than constant temperature, so the mean nuclear kinetic energy will fluctuate in the course of the simulation.
AIMD_THERMOSTAT
Applies thermostatting to AIMD trajectories.
TYPE:
INTEGER
DEFAULT:
none
OPTIONS:
LANGEVIN
Stochastic, white-noise Langevin thermostat
NOSE_HOOVER
Time-reversible, Nosé-Hoovery chain thermostat
RECOMMENDATION:
Use either thermostat for sampling the canonical (NVT) ensemble.
AIMD_TIME_STEP_CONVERSION
Modifies the molecular dynamics time step to increase granularity.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
The molecular dynamics time step is TIME_STEP/
RECOMMENDATION:
None
ANHAR_SEL
Select a subset of normal modes for subsequent anharmonic frequency analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use all normal modes
OPTIONS:
TRUE
Select subset of normal modes
RECOMMENDATION:
None
ANHAR
Performing various nuclear vibrational theory (TOSH, VPT2, VCI) calculations to obtain vibrational anharmonic frequencies.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Carry out the anharmonic frequency calculation.
FALSE
Do harmonic frequency calculation.
RECOMMENDATION:
Since this calculation involves the third and fourth derivatives at the minimum of the potential energy surface, it is recommended that the GEOM_OPT_TOL_DISPLACEMENT, GEOM_OPT_TOL_GRADIENT and GEOM_OPT_TOL_ENERGY tolerances are set tighter. Note that VPT2 calculations may fail if the system involves accidental degenerate resonances. See the VCI $rem variable for more details about increasing the accuracy of anharmonic calculations.
AO2MO_DISK
Sets the amount of disk space (in megabytes) available for MP2 calculations.
TYPE:
INTEGER
DEFAULT:
2000
Corresponding to 2000 Mb.
OPTIONS:
User-defined number of megabytes.
RECOMMENDATION:
Should be set as large as possible, discussed in Section 6.4.1.
ARI_R0
Determines the value of the inner fitting radius (in Ångstroms)
TYPE:
INTEGER
DEFAULT:
4
A value of 4 Å will be added to the atomic van der Waals radius.
OPTIONS:
User defined radius.
RECOMMENDATION:
For some systems the default value may be too small and the calculation will become unstable.
ARI_R1
Determines the value of the outer fitting radius (in Ångstroms)
TYPE:
INTEGER
DEFAULT:
5
A value of 5 Å will be added to the atomic van der Waals radius.
OPTIONS:
User defined radius.
RECOMMENDATION:
For some systems the default value may be too small and the calculation will become unstable. This value also determines, in part, the smoothness of the potential energy surface.
ARI
Toggles the use of the atomic resolution-of-the-identity (ARI) approximation.
TYPE:
LOGICAL
DEFAULT:
FALSE
ARI will not be used by default for an RI-JK calculation.
OPTIONS:
TRUE
Turn on ARI.
RECOMMENDATION:
For large (especially 1D and 2D) molecules the approximation may yield significant improvements in Fock evaluation time.
AUX_BASIS
Sets the auxiliary basis set to be used
TYPE:
STRING
DEFAULT:
No default auxiliary basis set
OPTIONS:
General, Gen
User-defined. As for BASIS
Symbol
Use standard auxiliary basis sets as in the table below
Mixed
Use a combination of different basis sets
RECOMMENDATION:
Consult literature and EMSL Basis Set Exchange to aid your selection.
BASIS2
Sets the small basis set to use in basis set projection.
TYPE:
STRING
DEFAULT:
No second basis set default.
OPTIONS:
Symbol. Use standard basis sets as per Chapter 8.
BASIS2_GEN
General BASIS2
BASIS2_MIXED
Mixed BASIS2
RECOMMENDATION:
BASIS2 should be smaller than BASIS. There is little advantage to using a basis larger than a minimal basis when BASIS2 is used for initial guess purposes. Larger, standardized BASIS2 options are available for dual-basis calculations (see Section 4.7).
BASISPROJTYPE
Determines which method to use when projecting the density matrix of BASIS2
TYPE:
STRING
DEFAULT:
FOPPROJECTION (when DUAL_BASIS_ENERGY=false)
OVPROJECTION (when DUAL_BASIS_ENERGY=true)
OPTIONS:
FOPPROJECTION
Construct the Fock matrix in the second basis
OVPROJECTION
Projects MOs from BASIS2 to BASIS.
RECOMMENDATION:
None
BASIS_LIN_DEP_THRESH
Sets the threshold for determining linear dependence in the basis set
TYPE:
INTEGER
DEFAULT:
6
Corresponding to a threshold of
OPTIONS:
Sets the threshold to
RECOMMENDATION:
Set to 5 or smaller if you have a poorly behaved SCF and you suspect linear dependence in you basis set. Lower values (larger thresholds) may affect the accuracy of the calculation.
BASIS
Specifies the basis sets to be used.
TYPE:
STRING
DEFAULT:
No default basis set
OPTIONS:
General, Gen
User defined ($basis keyword required).
Symbol
Use standard basis sets as per Chapter 8.
Mixed
Use a mixture of basis sets (see Chapter 8).
RECOMMENDATION:
Consult literature and reviews to aid your selection.
BOYSCALC
Specifies the Boys localized orbitals are to be calculated
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not perform localize the occupied space.
1
Allow core-valence mixing in Boys localization.
2
Localize core and valence separately.
RECOMMENDATION:
None
BOYS_CIS_NUMSTATE
Define how many states to mix with Boys localized diabatization. These states must be specified in the $localized_diabatization section.
TYPE:
INTEGER
DEFAULT:
0
Do not perform Boys localized diabatization.
OPTIONS:
2 to N where N is the number of CIS states requested (CIS_N_ROOTS)
RECOMMENDATION:
It is usually not wise to mix adiabatic states that are separated by more than a few eV or a typical reorganization energy in solvent.
CAGE_CHARGE
Defines the total charge of the cage.
TYPE:
INTEGER
DEFAULT:
400
Add a cage charged +4e.
OPTIONS:
Total charge of the cage is
RECOMMENDATION:
None
CAGE_POINTS
Defines number of point charges for the spherical cage.
TYPE:
INTEGER
DEFAULT:
100
OPTIONS:
Number of point charges to use.
RECOMMENDATION:
None
CAGE_RADIUS
Defines radius of the charged cage.
TYPE:
INTEGER
DEFAULT:
225
OPTIONS:
radius is
RECOMMENDATION:
None
CALC_NAC
Whether or not non-adiabatic couplings will be calculated for the EOM-CC, CIS, and TDDFT wave functions.
TYPE:
INTEGER
DEFAULT:
0 (do not compute NAC)
OPTIONS:
1
NYI for EOM-CC
2
Compute NACs using Szalay’s approach (this what needs to be specified for EOM-CC).
RECOMMENDATION:
Additional response equations will be solved and gradients for all EOM states and for summed states will be computed, which increases the cost of calculations. Request only when needed and do not ask for too many EOM states.
CALC_SOC
Whether or not the spin-orbit couplings between CC/EOM/ADC/CIS/TDDFT electronic states will be calculated. In the CC/EOM-CC suite, by default the couplings are calculated between the CCSD reference and the EOM-CCSD target states. In order to calculate couplings between EOM states, CC_STATE_TO_OPT must specify the initial EOM state.
TYPE:
LOGICAL
DEFAULT:
FALSE (no spin-orbit couplings will be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
One-electron and mean-field two-electron SOCs will be computed by default. To enable full two-electron SOCs, two-particle EOM properties must be turned on (see CC_EOM_PROP_TE).
CALC_SOC
Controls whether to calculate the SOC constants for EOM-CC, ADC, TDDFT/TDA and TDDFT.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform the SOC calculation.
TRUE
Perform the SOC calculation.
RECOMMENDATION:
None
CCVB_GUESS
Specifies the initial guess for CCVB calculations
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
1
Standard GVBMAN guess (orbital localization via GVB_LOCAL + Sano procedure).
2
Use orbitals from previous GVBMAN calculation, along with SCF_GUESS = read.
3
Convert UHF orbitals into pairing VB form.
RECOMMENDATION:
Option 1 is the most useful overall. The success of GVBMAN methods is often dependent on localized orbitals, and this guess shoots for these. Option 2 is useful for comparing results to other GVBMAN methods, or if other GVBMAN methods are able to obtain a desired result more efficiently. Option 3 can be useful for bond-breaking situations when a pertinent UHF solution has been found. It works best for small systems, or if the unrestriction is a local phenomenon within a larger molecule. If the unrestriction is non-local and the system is large, this guess will often produce a solution that is not the global minimum. Any UHF solution has a certain number of pairs that are unrestricted, and this will be output by the program. If GVB_N_PAIRS exceeds this number, the standard GVBMAN initial-guess procedure will be used to obtain a guess for the excess pairs
CCVB_METHOD
Optionally modifies the basic CCVB method
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Standard CCVB model
3
Independent electron pair approximation (IEPA) to CCVB
4
Variational PP (the CCVB reference energy)
RECOMMENDATION:
Option 1 is generally recommended. Option 4 is useful for preconditioning, and for obtaining localized-orbital solutions, which may be used in subsequent calculations. It is also useful for cases in which the regular GVBMAN PP code becomes variationally unstable. Option 3 is a simple independent-amplitude approximation to CCVB. It avoids the cubic-scaling amplitude equations of CCVB, and also is able to reach the correct dissociation energy for any molecular system (unlike regular CCVB which does so only for cases in which UHF can reach a correct dissociate limit). However the IEPA approximation to CCVB is sometimes variationally unstable, which we have yet to observe in regular CCVB.
CC_BACKEND
Used to specify the computational back-end of CCMAN2.
TYPE:
STRING
DEFAULT:
VM
Default shared-memory disk-based back-end
OPTIONS:
XM
libxm shared-memory disk-based back-end
CTF
Distributed-memory back-end for MPI jobs
RECOMMENDATION:
Use XM for large jobs with limited memory or when the performance of the default disk-based back-end is not satisfactory, CTF for MPI jobs
CC_CANONIZE_FINAL
Whether to semi-canonicalize orbitals at the end of the ground state calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
unless required
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Should not normally have to be altered.
CC_CANONIZE_FREQ
The orbitals will be semi-canonicalized every
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
User-defined integer
RECOMMENDATION:
Smaller values can be tried in cases that do not converge.
CC_CANONIZE
Whether to semi-canonicalize orbitals at the start of the calculation (i.e. Fock matrix is diagonalized in each orbital subspace)
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Should not normally have to be altered.
CC_CONVERGENCE
Overall convergence criterion for the coupled-cluster codes. This is designed to ensure at least
TYPE:
INTEGER
DEFAULT:
6
Energies.
7
Gradients.
OPTIONS:
Corresponding to
automatically to match energy convergence.
RECOMMENDATION:
Use the default
CC_DIIS12_SWITCH
When to switch from DIIS2 to DIIS1 procedure, or when DIIS2 procedure is required to generate DIIS guesses less frequently. Total value of DIIS error vector must be less than
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
User-defined integer
RECOMMENDATION:
None
CC_DIIS_FREQ
DIIS extrapolation will be attempted every n iterations. However, DIIS2 will be attempted every iteration while total error vector exceeds CC_DIIS12_SWITCH. DIIS1 cannot generate guesses more frequently than every 2 iterations.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:

User-defined integer
RECOMMENDATION:
None
CC_DIIS_MAX_OVERLAP
DIIS extrapolations will not begin until square root of the maximum element of the error overlap matrix drops below this value.
TYPE:
DOUBLE
DEFAULT:
100
Corresponding to 1.0
OPTIONS:
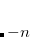
Integer code is mapped to
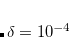
RECOMMENDATION:
None
CC_DIIS_MIN_OVERLAP
The DIIS procedure will be halted when the square root of smallest element of the error overlap matrix is less than
TYPE:
INTEGER
DEFAULT:
11
OPTIONS:
User-defined integer
RECOMMENDATION:
None
CC_DIIS_SIZE
Specifies the maximum size of the DIIS space.
TYPE:
INTEGER
DEFAULT:
7
OPTIONS:
User-defined integer
RECOMMENDATION:
Larger values involve larger amounts of disk storage.
CC_DIIS_START
Iteration number when DIIS is turned on. Set to a large number to disable DIIS.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
User-defined
RECOMMENDATION:
Occasionally DIIS can cause optimized orbital coupled-cluster calculations to diverge through large orbital changes. If this is seen, DIIS should be disabled.
CC_DIIS
Specify the version of Pulay’s Direct Inversion of the Iterative Subspace (DIIS) convergence accelerator to be used in the coupled-cluster code.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Activates procedure 2 initially, and procedure 1 when gradients are smaller
than DIIS12_SWITCH.
1
Uses error vectors defined as differences between parameter vectors from
successive iterations. Most efficient near convergence.
2
Error vectors are defined as gradients scaled by square root of the
approximate diagonal Hessian. Most efficient far from convergence.
RECOMMENDATION:
DIIS1 can be more stable. If DIIS problems are encountered in the early stages of a calculation (when gradients are large) try DIIS1.
CC_DIRECT_RI
Controls use of RI and Cholesky integrals in conventional (undecomposed) form
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
use all integrals in decomposed format
TRUE
transform all RI or Cholesky integral back to conventional format
RECOMMENDATION:
By default all integrals are used in decomposed format allowing significant reduction of memory use. If all integrals are transformed back (TRUE option) no memory reduction is achieved and decomposition error is introduced, however, the integral transformation is performed significantly faster and conventional CC/EOM algorithms are used.
CC_DOV_THRESH
Specifies minimum allowed values for the coupled-cluster energy denominators. Smaller values are replaced by this constant during early iterations only, so the final results are unaffected, but initial convergence is improved when the guess is poor.
TYPE:
INTEGER
DEFAULT:
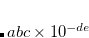
Corresponding to 0.25
OPTIONS:
Integer code is mapped to
RECOMMENDATION:
Increase to 0.5 or 0.75 for non convergent coupled-cluster calculations.
CC_DO_DYSON_EE
Whether excited-state or spin-flip state Dyson orbitals will be calculated for EOM-IP/EA-CCSD calculations with CCMAN.
TYPE:
LOGICAL
DEFAULT:
FALSE (the option must be specified to run this calculation)
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
none
CC_DO_DYSON
CCMAN2: starts all types of Dyson orbitals calculations. Desired type is determined by requesting corresponding EOM-XX transitions CCMAN: whether the reference-state Dyson orbitals will be calculated for EOM-IP/EA-CCSD calculations.
TYPE:
LOGICAL
DEFAULT:
FALSE (the option must be specified to run this calculation)
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
none
CC_EOM_2PA
Whether or not the transition moments and cross-sections for two-photon absorption will be calculated. By default, the transition moments are calculated between the CCSD reference and the EOM-CCSD target states. In order to calculate transition moments between a set of EOM-CCSD states and another EOM-CCSD state, the CC_STATE_TO_OPT must be specified for this state. If 2PA NTO analysis is requested, the CC_EOM_2PA value is redundant as long as CC_EOM_2PA
.
TYPE:
INTEGER
DEFAULT:
0 (do not compute 2PA transition moments)
OPTIONS:
1
Compute 2PA using the fastest algorithm (use
-intermediates for canonical
and
-intermediates for RI/CD response calculations).
2
Use
3
Use
RECOMMENDATION:
Additional response equations (6 for each target state) will be solved, which increases the cost of calculations. The cost of 2PA moments is about 10 times that of energy calculation. Use the default algorithm. Setting CC_EOM_2PA
CC_EOM_PROP
Whether or not the non-relaxed (expectation value) one-particle EOM-CCSD target state properties will be calculated. The properties currently include permanent dipole moment, the second moments
,
, and
of electron density, and the total
(in atomic units). Incompatible with JOBTYPE=FORCE, OPT, FREQ.
TYPE:
LOGICAL
DEFAULT:
FALSE (no one-particle properties will be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
Additional equations (EOM-CCSD equations for the left eigenvectors) need to be solved for properties, approximately doubling the cost of calculation for each irrep. The cost of the one-particle properties calculation itself is low. The one-particle density of an EOM-CCSD target state can be analyzed with NBO or libwfa packages by specifying the state with CC_STATE_TO_OPT and requesting NBO = TRUE and CC_EOM_PROP = TRUE.
CC_E_CONV
Convergence desired on the change in total energy, between iterations.
TYPE:
INTEGER
DEFAULT:
10
OPTIONS:
RECOMMENDATION:
None
CC_FNO_THRESH
Initialize the FNO truncation and sets the threshold to be used for both cutoffs (OCCT and POVO)
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
range
0000-10000
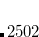
Corresponding to
%
RECOMMENDATION:
None
CC_FNO_USEPOP
Selection of the truncation scheme
TYPE:
INTEGER
DEFAULT:
1
OCCT
OPTIONS:
0
POVO
RECOMMENDATION:
None
CC_FULLRESPONSE
Fully relaxed properties (including orbital relaxation terms) will be computed. The variable CC_REF_PROP must be also set to TRUE.
TYPE:
LOGICAL
DEFAULT:
FALSE
(no orbital response will be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
Not available for non UHF/RHF references and for the methods that do not have analytic gradients (e.g., QCISD).
CC_HESS_THRESH
Minimum allowed value for the orbital Hessian. Smaller values are replaced by this constant.
TYPE:
DOUBLE
DEFAULT:
102
Corresponding to 0.01
OPTIONS:
Integer code is mapped to
RECOMMENDATION:
None
CC_INCL_CORE_CORR
Whether to include the correlation contribution from frozen core orbitals in non iterative (2) corrections, such as OD(2) and CCSD(2).
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
FALSE
RECOMMENDATION:
Use the default unless no core-valence or core correlation is desired (e.g., for comparison with other methods or because the basis used cannot describe core correlation).
CC_ITERATE_ON
In active space calculations, use a “mixed” iteration procedure if the value is greater than 0. Then if the RMS orbital gradient is larger than the value of CC_THETA_GRAD_THRESH, micro-iterations will be performed to converge the occupied-virtual mixing angles for the current active space. The maximum number of space iterations is given by this option.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Up to
RECOMMENDATION:
Can be useful for non-convergent active space calculations
CC_ITERATE_OV
In active space calculations, use a “mixed” iteration procedure if the value is greater than 0. Then, if the RMS orbital gradient is larger than the value of CC_THETA_GRAD_THRESH, micro-iterations will be performed to converge the occupied-virtual mixing angles for the current active space. The maximum number of such iterations is given by this option.
TYPE:
INTEGER
DEFAULT:
0
No “mixed” iterations
OPTIONS:
Up to
RECOMMENDATION:
Can be useful for non-convergent active space calculations.
CC_MAX_ITER
Maximum number of iterations to optimize the coupled-cluster energy.
TYPE:
INTEGER
DEFAULT:
200
OPTIONS:
up to
RECOMMENDATION:
None
CC_MEMORY
Specifies the maximum size, in Mb, of the buffers for in-core storage of block-tensors in CCMAN and CCMAN2.
TYPE:
INTEGER
DEFAULT:
50% of MEM_TOTAL. If MEM_TOTAL is not set, use 1.5 Gb. A minimum of
192 Mb is hard-coded.
OPTIONS:
Integer number of Mb
RECOMMENDATION:
Larger values can give better I/O performance and are recommended for systems with large memory (add to your .qchemrc file. When running CCMAN2 exclusively on a node, CC_MEMORY should be set to 75–80% of the total available RAM. )
CC_MP2NO_GRAD
If CC_MP2NO_GUESS is TRUE, what kind of one-particle density matrix is used to make the guess orbitals?
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
1 PDM from MP2 gradient theory.
FALSE
1 PDM expanded to 2
order in perturbation theory.
RECOMMENDATION:
The two definitions give generally similar performance.
CC_MP2NO_GUESS
Will guess orbitals be natural orbitals of the MP2 wave function? Alternatively, it is possible to use an effective one-particle density matrix to define the natural orbitals.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Use natural orbitals from an MP2 one-particle density matrix (see CC_MP2NO_GRAD).
FALSE
Use current molecular orbitals from SCF.
RECOMMENDATION:
None
CC_ORBS_PER_BLOCK
Specifies target (and maximum) size of blocks in orbital space.
TYPE:
INTEGER
DEFAULT:
16
OPTIONS:
Orbital block size of
RECOMMENDATION:
None
CC_POL
Whether or not the static polarizability for the CCSD wave function will be calculated.
TYPE:
LOGICAL
DEFAULT:
FALSE (CCSD static polarizability will not be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
Static polarizabilities are expensive since they require solving three additional response equations. Do no request this property unless you need it.
CC_PRECONV_FZ
In active space methods, whether to pre-converge other wave function variables for fixed initial guess of active space.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No pre-iterations before active space optimization begins.
Maximum number of pre-iterations via this procedure.
RECOMMENDATION:
None
CC_PRECONV_T2Z_EACH
Whether to pre-converge the cluster amplitudes before each change of the orbitals in optimized orbital coupled-cluster methods. The maximum number of iterations in this pre-convergence procedure is given by the value of this parameter.
TYPE:
INTEGER
DEFAULT:
0
(FALSE)
OPTIONS:
0
No pre-convergence before orbital optimization.
Up to
RECOMMENDATION:
A very slow last resort option for jobs that do not converge.
CC_PRECONV_T2Z
Whether to pre-converge the cluster amplitudes before beginning orbital optimization in optimized orbital cluster methods.
TYPE:
INTEGER
DEFAULT:
0
(FALSE)
10
If CC_RESTART, CC_RESTART_NO_SCF or CC_MP2NO_GUESS are TRUE
OPTIONS:
0
No pre-convergence before orbital optimization.
Up to
RECOMMENDATION:
Experiment with this option in cases of convergence failure.
CC_PRINT
Controls the output from post-MP2 coupled-cluster module of Q-Chem
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
higher values can lead to deforestation…
RECOMMENDATION:
Increase if you need more output and don’t like trees
CC_QCCD_THETA_SWITCH
QCCD calculations switch from OD to QCCD when the rotation gradient is below this threshold [
TYPE:
INTEGER
DEFAULT:
2
switchover
OPTIONS:
RECOMMENDATION:
None
CC_REF_PROP_TE
Request for calculation of non-relaxed two-particle CCSD properties. The two-particle properties currently include
. The one-particle properties also will be calculated, since the additional cost of the one-particle properties calculation is inferior compared to the cost of
TYPE:
LOGICAL
DEFAULT:
FALSE
(no two-particle properties will be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
The two-particle properties are computationally expensive, since they require calculation and use of the two-particle density matrix (the cost is approximately the same as the cost of an analytic gradient calculation). Do not request the two-particle properties unless you really need them.
CC_REF_PROP
Whether or not the non-relaxed (expectation value) or full response (including orbital relaxation terms) one-particle CCSD properties will be calculated. The properties currently include permanent dipole moment, the second moments
TYPE:
LOGICAL
DEFAULT:
FALSE
(no one-particle properties will be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
Additional equations need to be solved (lambda CCSD equations) for properties with the cost approximately the same as CCSD equations. Use the default if you do not need properties. The cost of the properties calculation itself is low. The CCSD one-particle density can be analyzed with NBO package by specifying NBO=TRUE, CC_REF_PROP=TRUE and JOBTYPE=FORCE.
CC_RESET_THETA
The reference MO coefficient matrix is reset every n iterations to help overcome problems associated with the theta metric as theta becomes large.
TYPE:
INTEGER
DEFAULT:
15
OPTIONS:
RECOMMENDATION:
None
CC_RESTART_NO_SCF
Should an optimized orbital coupled cluster calculation begin with optimized orbitals from a previous calculation? When TRUE, molecular orbitals are initially orthogonalized, and CC_PRECONV_T2Z and CC_CANONIZE are set to TRUE while other guess options are set to FALSE
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
None
CC_RESTART
Allows an optimized orbital coupled cluster calculation to begin with an initial guess for the orbital transformation matrix U other than the unit vector. The scratch file from a previous run must be available for the U matrix to be read successfully.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Use unit initial guess.
TRUE
Activates CC_PRECONV_T2Z, CC_CANONIZE, and
turns off CC_MP2NO_GUESS
RECOMMENDATION:
Useful for restarting a job that did not converge, if files were saved.
CC_RESTR_AMPL
Controls the restriction on amplitudes is there are restricted orbitals
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
0
All amplitudes are in the full space
1
Amplitudes are restricted, if there are restricted orbitals
RECOMMENDATION:
None
CC_RESTR_TRIPLES
Controls which space the triples correction is computed in
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Triples are computed in the full space
1
Triples are restricted to the active space
RECOMMENDATION:
None
CC_REST_AMPL
Forces the integrals,
, and
amplitudes to be determined in the full space even though the CC_REST_OCC and CC_REST_VIR keywords are used.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
FALSE
Do apply restrictions
TRUE
Do not apply restrictions
RECOMMENDATION:
None
CC_REST_OCC
Sets the number of restricted occupied orbitals including active core occupied orbitals.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Restrict
RECOMMENDATION:
Example: cytosine with the molecular formula C
H
N
O includes one oxygen atom. To calculate O 1s core-excited states,
CC_REST_TRIPLES
Restricts
amplitudes to the active space, i.e., one electron should be removed from the active occupied orbital and one electron should be added to the active virtual orbital.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Applies the restrictions
RECOMMENDATION:
None
CC_REST_VIR
Sets the number of restricted virtual orbitals including frozen virtual orbitals.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Restrict
RECOMMENDATION:
None
CC_SCALE_AMP
If not 0, scales down the step for updating coupled-cluster amplitudes in cases of problematic convergence.
TYPE:
INTEGER
DEFAULT:
0
no scaling
OPTIONS:
Integer code is mapped to
, e.g.,
corresponds to 0.9
RECOMMENDATION:
Use 0.9 or 0.8 for non convergent coupled-cluster calculations.
CC_STATE_TO_OPT
Specifies which state to optimize.
TYPE:
INTEGER ARRAY
DEFAULT:
None
OPTIONS:
[
,
]
optimize the
RECOMMENDATION:
None
CC_SYMMETRY
Activates point-group symmetry in the ADC calculation.
TYPE:
LOGICAL
DEFAULT:
TRUE
If the system possesses any point-group symmetry.
OPTIONS:
TRUE
Employ point-group symmetry
FALSE
Do not use point-group symmetry
RECOMMENDATION:
None
CC_THETA_CONV
Convergence criterion on the RMS difference between successive sets of orbital rotation angles [
TYPE:
INTEGER
DEFAULT:
5
Energies
6
Gradients
OPTIONS:
RECOMMENDATION:
Use default
CC_THETA_GRAD_CONV
Convergence desired on the RMS gradient of the energy with respect to orbital rotation angles [
TYPE:
INTEGER
DEFAULT:
7
Energies
8
Gradients
OPTIONS:
RECOMMENDATION:
Use default
CC_THETA_GRAD_THRESH
RMS orbital gradient threshold [
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
RECOMMENDATION:
Can be made smaller if convergence difficulties are encountered.
CC_THETA_STEPSIZE
Scale factor for the orbital rotation step size. The optimal rotation steps should be approximately equal to the gradient vector.
TYPE:
INTEGER
DEFAULT:
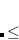
Corresponding to 1.0
OPTIONS:
Integer code is mapped to
If the initial step is smaller than 0.5, the program will increase step
when gradients are smaller than the value of THETA_GRAD_THRESH,
up to a limit of 0.5.
RECOMMENDATION:
Try a smaller value in cases of poor convergence and very large orbital gradients. For example, a value of 01001 translates to 0.1
CC_TRANS_PROP
Whether or not the transition dipole moment (in atomic units) and oscillator strength for the EOM-CCSD target states will be calculated. By default, the transition dipole moment is calculated between the CCSD reference and the EOM-CCSD target states. In order to calculate transition dipole moment between a set of EOM-CCSD states and another EOM-CCSD state, the CC_STATE_TO_OPT must be specified for this state.
TYPE:
INTEGER
DEFAULT:
0 (no transition properties will be calculated)
OPTIONS:
1 (calculate transition properties between all computed EOM state and the reference state)
2 (calculate transition properties between all pairs of EOM states)
RECOMMENDATION:
NONE
Additional equations (for the left EOM-CCSD eigenvectors plus lambda CCSD equations in case if transition properties between the CCSD reference and EOM-CCSD target states are requested) need to be solved for transition properties, approximately doubling the computational cost. The cost of the transition properties calculation itself is low.
CC_T_CONV
Convergence criterion on the RMS difference between successive sets of coupled-cluster doubles amplitudes [
TYPE:
INTEGER
DEFAULT:
8
energies
10
gradients
OPTIONS:
RECOMMENDATION:
Use default
CC_Z_CONV
Convergence criterion on the RMS difference between successive doubles
-vector amplitudes [
TYPE:
INTEGER
DEFAULT:
8
Energies
10
Gradients
OPTIONS:
RECOMMENDATION:
Use Default
CDFTCI_PRINT
Controls level of output from CDFT-CI procedure to Q-Chem output file.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Only print energies and coefficients of CDFT-CI final states
1
Level 0 plus CDFT-CI overlap, Hamiltonian, and population matrices
2
Level 1 plus eigenvectors and eigenvalues of the CDFT-CI population matrix
3
Level 2 plus promolecule orbital coefficients and energies
RECOMMENDATION:
Level 3 is primarily for program debugging; levels 1 and 2 may be useful for analyzing the coupling elements
CDFTCI_RESTART
To be used in conjunction with CDFTCI_STOP, this variable causes CDFT-CI to read already-converged states from disk and begin SCF convergence on later states. Note that the same $cdft section must be used for the stopped calculation and the restarted calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Start calculations on state
RECOMMENDATION:
Use this setting in conjunction with CDFTCI_STOP.
CDFTCI_SKIP_PROMOLECULES
Skips promolecule calculations and allows fractional charge and spin constraints to be specified directly.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Standard CDFT-CI calculation is performed.
TRUE
Use the given charge/spin constraints directly, with no promolecule calculations.
RECOMMENDATION:
Setting to TRUE can be useful for scanning over constraint values.
CDFTCI_STOP
The CDFT-CI procedure involves performing independent SCF calculations on distinct constrained states. It sometimes occurs that the same convergence parameters are not successful for all of the states of interest, so that a CDFT-CI calculation might converge one of these diabatic states but not the next. This variable allows a user to stop a CDFT-CI calculation after a certain number of states have been converged, with the ability to restart later on the next state, with different convergence options.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Stop after converging state
)
Do not stop early
RECOMMENDATION:
Use this setting if some diabatic states converge but others do not.
CDFTCI_SVD_THRESH
By default, a symmetric orthogonalization is performed on the CDFT-CI matrix before diagonalization. If the CDFT-CI overlap matrix is nearly singular (i.e., some of the diabatic states are nearly degenerate), then this orthogonalization can lead to numerical instability. When computing
, eigenvalues smaller than
are discarded.
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
for a threshold of
RECOMMENDATION:
Can be decreased if numerical instabilities are encountered in the final diagonalization.
CDFTCI
Initiates a constrained DFT-configuration interaction calculation
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform a CDFT-CI Calculation
FALSE
No CDFT-CI
RECOMMENDATION:
Set to TRUE if a CDFT-CI calculation is desired.
CDFT_BECKE_POP
Whether the calculation should print the Becke atomic charges at convergence
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Print Populations
FALSE
Do not print them
RECOMMENDATION:
Use the default. Note that the Mulliken populations printed at the end of an SCF run will not typically add up to the prescribed constraint value. Only the Becke populations are guaranteed to satisfy the user-specified constraints.
CDFT_CRASHONFAIL
Whether the calculation should crash or not if the constraint iterations do not converge.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Crash if constraint iterations do not converge.
FALSE
Do not crash.
RECOMMENDATION:
Use the default.
CDFT_LAMBDA_MODE
Allows CDFT potentials to be specified directly, instead of being determined as Lagrange multipliers.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Standard CDFT calculations are used.
TRUE
Instead of specifying target charge and spin constraints, use the values
from the input deck as the value of the Becke weight potential
RECOMMENDATION:
Should usually be set to FALSE. Setting to TRUE can be useful to scan over different strengths of charge or spin localization, as convergence properties are improved compared to regular CDFT(-CI) calculations.
CDFT_POP
Sets the charge partitioning scheme for cDFT in SAPT/cDFT
TYPE:
STRING
DEFAULT:
FBH
OPTIONS:
FBH
Fragment-Based Hirshfeld partitioning
BECKE
Atomic Becke partitioning
RECOMMENDATION:
None
CDFT_POSTDIIS
Controls whether the constraint is enforced after DIIS extrapolation.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Enforce constraint after DIIS
FALSE
Do not enforce constraint after DIIS
RECOMMENDATION:
Use the default unless convergence problems arise, in which case it may be beneficial to experiment with setting CDFT_POSTDIIS to FALSE. With this option set to TRUE, energies should be variational after the first iteration.
CDFT_PREDIIS
Controls whether the constraint is enforced before DIIS extrapolation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Enforce constraint before DIIS
FALSE
Do not enforce constraint before DIIS
RECOMMENDATION:
Use the default unless convergence problems arise, in which case it may be beneficial to experiment with setting CDFT_PREDIIS to TRUE. Note that it is possible to enforce the constraint both before and after DIIS by setting both CDFT_PREDIIS and CDFT_POSTDIIS to TRUE.
CDFT_THRESH
Threshold that determines how tightly the constraint must be satisfied.
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
N
Constraint is satisfied to within
.
RECOMMENDATION:
Use the default unless problems occur.
CDFT
Initiates a constrained DFT calculation
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform a Constrained DFT Calculation
FALSE
No Density Constraint
RECOMMENDATION:
Set to TRUE if a Constrained DFT calculation is desired.
CD_ALGORITHM
Determines the algorithm for MP2 integral transformations.
TYPE:
STRING
DEFAULT:
Program determined.
OPTIONS:
DIRECT
Uses fully direct algorithm (energies only).
SEMI_DIRECT
Uses disk-based semi-direct algorithm.
LOCAL_OCCUPIED
Alternative energy algorithm (see 6.4.1).
RECOMMENDATION:
Semi-direct is usually most efficient, and will normally be chosen by default.
CFMM_ORDER
Controls the order of the multipole expansions in CFMM calculation.
TYPE:
INTEGER
DEFAULT:
15
For single point SCF accuracy
25
For tighter convergence (optimizations)
OPTIONS:
Use multipole expansions of order
RECOMMENDATION:
Use the default.
CHARGE_CHARGE_REPULSION
The repulsive Coulomb interaction parameter for YinYang atoms.
TYPE:
INTEGER
DEFAULT:
550
OPTIONS:
Use Q =

RECOMMENDATION:
The repulsive Coulomb potential maintains bond lengths involving YinYang atoms with the potential
. The default is parameterized for carbon atoms.
CHELPG_DX
Sets the rectangular grid spacing for the traditional Cartesian ChElPG grid or the spacing between concentric Lebedev shells (when the variables CHELPG_HA and CHELPG_H are specified as well).
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
Corresponding to a grid space of
, in Å.
RECOMMENDATION:
Use the default, which corresponds to the “dense grid” of Breneman and Wiberg,[Breneman and Wiberg(1990)], unless the cost is prohibitive, in which case a larger value can be selected. Note that this default value is set with the Cartesian grid in mind and not the Lebedev grid. In the Lebedev case, a larger value can typically be used.
CHELPG_HA
Sets the Lebedev grid to use for non-hydrogen atoms.
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
Corresponding to a number of points in a Lebedev grid (see Section 5.5.1.
RECOMMENDATION:
None.
CHELPG_HEAD
Sets the “head space”[Breneman and Wiberg(1990)] (radial extent) of the ChElPG grid.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
Corresponding to a head space of
, in Å.
RECOMMENDATION:
Use the default, which is the value recommended by Breneman and Wiberg.[Breneman and Wiberg(1990)]
CHELPG_H
Sets the Lebedev grid to use for hydrogen atoms.
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
Corresponding to a number of points in a Lebedev grid.
RECOMMENDATION:
CHELPG_H must always be less than or equal to CHELPG_HA. If it is greater, it will automatically be set to the value of CHELPG_HA.
CHELPG
Controls the calculation of CHELPG charges.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not calculate ChElPG charges.
TRUE
Compute ChElPG charges.
RECOMMENDATION:
Set to TRUE if desired. For large molecules, there is some overhead associated with computing ChElPG charges, especially if the number of grid points is large.
CHILD_MP_ORDERS
The multipole orders included in the prepared FERFs. The last digit specifies how many multipoles to compute, and the digits in the front specify the multipole orders: 2: dipole (D); 3: quadrupole (Q); 4: octopole (O).
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
21
D
232
DQ
2343
DQO
RECOMMENDATION:
Use 232 (DQ) when FERF is needed.
CHILD_MP
Compute FERFs for fragments and use them as the basis for SCFMI calculations.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not compute FERFs (use the full AO span of each fragment).
TRUE
Compute fragment FERFs.
RECOMMENDATION:
Use FERFs to compute polarization energy when large basis sets are used. In an “EDA2" calculation, this $rem variable is set based on the given option automatically.
CHOLESKY_TOL
Tolerance of Cholesky decomposition of two-electron integrals
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
Corresponds to a tolerance of
RECOMMENDATION:
2 - qualitative calculations, 3 - appropriate for most cases, 4 - quantitative (error in total energy typically less than 1
CISTR_PRINT
Controls level of output.
TYPE:
LOGICAL
DEFAULT:
FALSE
Minimal output.
OPTIONS:
TRUE
Increase output level.
RECOMMENDATION:
None
CIS_AMPL_ANAL
Perform additional analysis of CIS and TDDFT excitation amplitudes, including generation of natural transition orbitals, excited-state multipole moments, and Mulliken analysis of the excited state densities and particle/hole density matrices.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform additional amplitude analysis.
FALSE
Do not perform additional analysis.
RECOMMENDATION:
None
CIS_CONVERGENCE
CIS is considered converged when error is less than
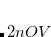
TYPE:
INTEGER
DEFAULT:
6
CIS convergence threshold 10
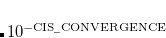
OPTIONS:
Corresponding to
RECOMMENDATION:
None
CIS_DER_NUMSTATE
Determines among how many states we calculate non-adiabatic couplings. These states must be specified in the $derivative_coupling section.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not calculate non-adiabatic couplings.
Calculate
pairs of non-adiabatic couplings.
RECOMMENDATION:
None.
CIS_DIABATH_DECOMPOSE
Decide whether or not to decompose the diabatic coupling into Coulomb, exchange, and one-electron terms.
TYPE:
LOGICAL
DEFAULT:
FALSE
Do not decompose the diabatic coupling.
OPTIONS:
TRUE
RECOMMENDATION:
These decompositions are most meaningful for electronic excitation transfer processes. Currently, available only for CIS, not for TDDFT diabatic states.
CIS_DYNAMIC_MEM
Controls whether to use static or dynamic memory in CIS and TDDFT calculations.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Partly use static memory
TRUE
Fully use dynamic memory
RECOMMENDATION:
The default control requires static memory (MEM_STATIC) to hold a temporary array whose minimum size is
. For a large calculation, one has to specify a large value for MEM_STATIC, which is not recommended (see Chapter 2). Therefore, it is recommended to use dynamic memory for large calculations.
CIS_GUESS_DISK_TYPE
Determines the type of guesses to be read from disk
TYPE:
INTEGER
DEFAULT:
Nil
OPTIONS:
0
Read triplets only
1
Read triplets and singlets
2
Read singlets only
RECOMMENDATION:
Must be specified if CIS_GUESS_DISK is TRUE.
CIS_GUESS_DISK
Read the CIS guess from disk (previous calculation).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Create a new guess.
TRUE
Read the guess from disk.
RECOMMENDATION:
Requires a guess from previous calculation.
CIS_MOMENTS
Controls calculation of excited-state (CIS or TDDFT) multipole moments
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not calculate excited-state moments.
TRUE
Calculate moments for each excited state.
RECOMMENDATION:
Set to TRUE if excited-state moments are desired. (This is a trivial additional calculation.) The MULTIPOLE_ORDER controls how many multipole moments are printed.
CIS_MULLIKEN
Controls Mulliken and Löwdin population analyses for excited-state particle and hole density matrices.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform particle/hole population analysis.
TRUE
Perform both Mulliken and Löwdin analysis of the particle and hole
density matrices for each excited state.
RECOMMENDATION:
Set to TRUE if desired. This represents a trivial additional calculation.
CIS_N_ROOTS
Sets the number of excited state roots to find
TYPE:
INTEGER
DEFAULT:
0
Do not look for any excited states
OPTIONS:
Looks for
RECOMMENDATION:
None
CIS_RELAXED_DENSITY
Use the relaxed CIS density for attachment/detachment density analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not use the relaxed CIS density in analysis.
TRUE
Use the relaxed CIS density in analysis.
RECOMMENDATION:
None
CIS_S2_THRESH
Determines whether a state is a singlet or triplet in unrestricted calculations.
TYPE:
INTEGER
DEFAULT:
120
OPTIONS:
Sets the
threshold to

RECOMMENDATION:
For the default case, states with
are treated as triplet states and other states are treated as singlets.
CIS_SINGLETS
Solve for singlet excited states (ignored for spin unrestricted systems)
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Solve for singlet states
FALSE
Do not solve for singlet states.
RECOMMENDATION:
None
CIS_STATE_DERIV
Sets CIS state for excited state optimizations and vibrational analysis.
TYPE:
INTEGER
DEFAULT:
0
Does not select any of the excited states.
OPTIONS:
Select the
RECOMMENDATION:
Check to see that the states do not change order during an optimization, due to state crossings.
CIS_TRIPLETS
Solve for triplet excited states (ignored for spin unrestricted systems)
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Solve for triplet states
FALSE
Do not solve for triplet states.
RECOMMENDATION:
None
CM5
Controls running of CM5 population analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculate CM5 populations.
FALSE
Do not calculate CM5 populations.
RECOMMENDATION:
None
COMBINE_K
Controls separate or combined builds for short-range and long-range K
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Build short-range and long-range K separately (twice as expensive as a global hybrid)
TRUE (or 1)
Build short-range and long-range K together (
as expensive as a global hybrid)
RECOMMENDATION:
Most pre-defined range-separated hybrid functionals in Q-Chem use this feature by default. However, if a user-specified RSH is desired, it is necessary to manually turn this feature on.
COMPLEX_CCMAN
Requests complex-scaled or CAP-augmented CC/EOM calculations.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Engage complex CC/EOM code.
RECOMMENDATION:
Not available in CCMAN. Need to specify CAP strength or complex-scaling parameter in $complex_ccman section.
COMPLEX_MIX
Mix a certain percentage of the real part of the HOMO to the imaginary part of the LUMO.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0–100
The mix angle =
COMPLEX_MIX/100.
RECOMMENDATION:
It may help find the stable complex solution (similar idea as SCF_GUESS_MIX).
COMPLEX
Run an SCF calculation with complex MOs using GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Use complex orbitals.
FALSE
Use real orbitals.
RECOMMENDATION:
Set to TRUE if desired.
CORE_CHARACTER
Selects how the core orbitals are determined in the frozen-core approximation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use energy-based definition.
1-4
Use Mulliken-based definition (see Table 6.2 for details).
RECOMMENDATION:
Use the default, unless performing calculations on molecules with heavy elements.
CORE_IONIZE
Indicates how orbitals are specified for reduced excitation spaces.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
all valence orbitals are listed in $solute section
2
only hole(s) are specified all other occupations same as ground state
RECOMMENDATION:
For MOM + TDDFT this specifies the input form of the $solute section. If set to 1 all occupied orbitals must be specified, 2 only the empty orbitals to ignore must be specified.
CORRELATION
Specifies the correlation level of theory handled by CCMAN/CCMAN2.
TYPE:
STRING
DEFAULT:
None
No Correlation
OPTIONS:
CCMP2
Regular MP2 handled by CCMAN/CCMAN2
MP3
CCMAN and CCMAN2
MP4SDQ
CCMAN
MP4
CCMAN
CCD
CCMAN and CCMAN2
CCD(2)
CCMAN
CCSD
CCMAN and CCMAN2
CCSD(T)
CCMAN and CCMAN2
CCSD(2)
CCMAN
CCSD(fT)
CCMAN and CCMAN2
CCSD(dT)
CCMAN
CCVB-SD
CCMAN2
QCISD
CCMAN and CCMAN2
QCISD(T)
CCMAN and CCMAN2
OD
CCMAN
OD(T)
CCMAN
OD(2)
CCMAN
VOD
CCMAN
VOD(2)
CCMAN
QCCD
CCMAN
QCCD(T)
CCMAN
QCCD(2)
CCMAN
VQCCD
CCMAN
VQCCD(T)
CCMAN
VQCCD(2)
CCMAN
RECOMMENDATION:
Consult the literature for guidance.
CPSCF_NSEG
Controls the number of segments used to calculate the CPSCF equations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not solve the CPSCF equations in segments.
User-defined. Use
RECOMMENDATION:
Use the default.
CUBEFILE_STATE
Determines which excited state is used to generate cube files
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
Generate cube files for the
RECOMMENDATION:
None
CUDA_RI-MP2
Enables GPU implementation of RI-MP2
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
GPU-enabled MGEMM off
TRUE
GPU-enabled MGEMM on
RECOMMENDATION:
Necessary to set to 1 in order to run GPU-enabled RI-MP2
CUTOCC
Specifies occupied orbital cutoff.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
0-200
CUTOFF = CUTOCC/100
RECOMMENDATION:
None
CUTVIR
Specifies virtual orbital cutoff.
TYPE:
INTEGER
DEFAULT:
0
No truncation
OPTIONS:
0-100
CUTOFF = CUTVIR/100
RECOMMENDATION:
None
DEUTERATE
Requests that all hydrogen atoms be replaces with deuterium.
TYPE:
LOGICAL
DEFAULT:
FALSE
Do not replace hydrogens.
OPTIONS:
TRUE
Replace hydrogens with deuterium.
RECOMMENDATION:
Replacing hydrogen atoms reduces the fastest vibrational frequencies by a factor of 1.4, which allow for a larger fictitious mass and time step in ELMD calculations. There is no reason to replace hydrogens in BOMD calculations.
DFPT_EXCHANGE
Specifies the secondary functional in a HFPC/DFPC calculation.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
None
RECOMMENDATION:
See reference for recommended basis set, functional, and grid pairings.
DFPT_XC_GRID
Specifies the secondary grid in a HFPC/DFPC calculation.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
None
RECOMMENDATION:
See reference for recommended basis set, functional, and grid pairings.
DFTVDW_ALPHA1
Parameter in XDM calculation with higher-order terms
TYPE:
INTEGER
DEFAULT:
83
OPTIONS:
10-1000
RECOMMENDATION:
None
DFTVDW_ALPHA2
Parameter in XDM calculation with higher-order terms.
TYPE:
INTEGER
DEFAULT:
155
OPTIONS:
10-1000
RECOMMENDATION:
None
DFTVDW_JOBNUMBER
Basic vdW job control
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not apply the XDM scheme.
1
Add vdW as energy/gradient correction to SCF.
2
Add vDW as a DFT functional and do full SCF (this option only works with XDM6).
RECOMMENDATION:
None
DFTVDW_KAI
Damping factor
for
-only damping function
TYPE:
INTEGER
DEFAULT:
800
OPTIONS:
10–1000
RECOMMENDATION:
None
DFTVDW_METHOD
Choose the damping function used in XDM
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Use Becke’s damping function including
2
Use Becke’s damping function with higher-order (
and
) terms.
RECOMMENDATION:
None
DFTVDW_MOL1NATOMS
The number of atoms in the first monomer in dimer calculation
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0–
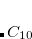
RECOMMENDATION:
None
DFTVDW_PRINT
Printing control for VDW code
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
0
No printing.
1
Minimum printing (default)
2
Debug printing
RECOMMENDATION:
None
DFTVDW_USE_ELE_DRV
Specify whether to add the gradient correction to the XDM energy. only valid with Becke’s
TYPE:
LOGICAL
DEFAULT:
1
OPTIONS:
1
Use density correction when applicable.
0
Do not use this correction (for debugging purposes).
RECOMMENDATION:
None
DFT_C
Controls whether the DFT-C empirical BSSE correction should be added.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
(or 0) Do not apply the DFT-C correction
TRUE
(or 1) Apply the DFT-C correction
RECOMMENDATION:
NONE
DFT_D3_3BODY
Controls whether the three-body interaction in Grimme’s DFT-D3 method should be applied (see Eq. (14) in Ref. Grimme:2010).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Do not apply the three-body interaction term
TRUE
Apply the three-body interaction term
RECOMMENDATION:
NONE
DFT_D3_A1
The nonlinear parameter
in Eqs. eq:bj-damping, eq:cso-damping, eq:zero-m-damping, and eq:op-damping. Used in DFT-D3(BJ), DFT-D3(CSO), DFT-D3M(0), DFT-D3M(BJ), and DFT-D3(op).
TYPE:
INTEGER
DEFAULT:
100000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D3_A2
The nonlinear parameter
in Eqs. eq:bj-damping and eq:op-damping. Used in DFT-D3(BJ), DFT-D3M(BJ), and DFT-D3(op).
TYPE:
INTEGER
DEFAULT:
100000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D3_POWER
The nonlinear parameter
in Eq. eq:op-damping. Used in DFT-D3(op). Must be greater than or equal to 6 to avoid divergence.
TYPE:
INTEGER
DEFAULT:
600000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D3_RS6
The nonlinear parameter
in Eqs. eq:zero-damping and Eq. eq:zero-m-damping. Used in DFT-D3(0) and DFT-D3M(0).
TYPE:
INTEGER
DEFAULT:
100000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D3_RS8
The nonlinear parameter
in Eqs. eq:zero-damping and Eq. eq:zero-m-damping. Used in DFT-D3(0) and DFT-D3M(0).
TYPE:
INTEGER
DEFAULT:
100000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D3_S6
The linear parameter
in eq. (5.32). Used in all forms of DFT-D3.
TYPE:
INTEGER
DEFAULT:
100000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D3_S8
The linear parameter
in Eq. eq:D3eqn. Used in DFT-D3(0), DFT-D3(BJ), DFT-D3M(0), DFT-D3M(BJ), and DFT-D3(op).
TYPE:
INTEGER
DEFAULT:
100000
OPTIONS:
Corresponding to
.
RECOMMENDATION:
NONE
DFT_D_A
Controls the strength of dispersion corrections in the Chai–Head-Gordon DFT-D scheme, Eq. eqn:CHG.
TYPE:
INTEGER
DEFAULT:
600
OPTIONS:
Corresponding to
.
RECOMMENDATION:
Use the default.
DFT_D
Controls the empirical dispersion correction to be added to a DFT calculation.
TYPE:
LOGICAL
DEFAULT:
None
OPTIONS:
FALSE
(or 0) Do not apply the DFT-D2, DFT-CHG, or DFT-D3 scheme
EMPIRICAL_GRIMME
DFT-D2 dispersion correction from Grimme[Grimme(2006b)]
EMPIRICAL_CHG
DFT-CHG dispersion correction from Chai and Head-Gordon[Chai and Head-Gordon(2008b)]
EMPIRICAL_GRIMME3
DFT-D3(0) dispersion correction from Grimme (deprecated as
of Q-Chem 5.0)
D3_ZERO
DFT-D3(0) dispersion correction from Grimme et al.[Grimme et al.(2010)Grimme, Antony, Ehrlich, and Krieg]
D3_BJ
DFT-D3(BJ) dispersion correction from Grimme et al.[Grimme et al.(2011)Grimme, Ehrlich, and Goerigk]
D3_CSO
DFT-D3(CSO) dispersion correction from Schröder et al.[Schröder et al.(2015)Schröder, Creon, and Schwabe]
D3_ZEROM
DFT-D3M(0) dispersion correction from Smith et al.[Smith et al.(2016)Smith, Burns, Patkowski, and Sherrill]
D3_BJM
DFT-D3M(BJ) dispersion correction from Smith et al.[Smith et al.(2016)Smith, Burns, Patkowski, and Sherrill]
D3_OP
DFT-D3(op) dispersion correction from Witte et al.[Witte et al.(2017a)Witte, Mardirossian, Neaton, and Head-Gordon]
D3
Automatically select the "best" available D3 dispersion correction
RECOMMENDATION:
Use the D3 option, which selects the empirical potential based on the density functional specified by the user.
DH
Controls the application of DH-DFT scheme.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Do not apply the DH-DFT scheme
TRUE (or 1)
Apply DH-DFT scheme
RECOMMENDATION:
NONE
DIIS_ERR_RMS
Changes the DIIS convergence metric from the maximum to the RMS error.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE, FALSE
RECOMMENDATION:
Use the default, the maximum error provides a more reliable criterion.
DIIS_PRINT
Controls the output from DIIS SCF optimization.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Minimal print out.
1
Chosen method and DIIS coefficients and solutions.
2
Level 1 plus changes in multipole moments.
3
Level 2 plus Multipole moments.
4
Level 3 plus extrapolated Fock matrices.
RECOMMENDATION:
Use the default
DIIS_SEPARATE_ERRVEC
Control optimization of DIIS error vector in unrestricted calculations.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use a combined
and
error vector.
OPTIONS:
FALSE
Use a combined
TRUE
Use separate error vectors for the
RECOMMENDATION:
When using DIIS in Q-Chem a convenient optimization for unrestricted calculations is to sum the
DIIS_SUBSPACE_SIZE
Controls the size of the DIIS and/or RCA subspace during the SCF.
TYPE:
INTEGER
DEFAULT:
15
OPTIONS:
User-defined
RECOMMENDATION:
None
DIP_SINGLETS
Sets the number of singlet DIP roots to find. Valid only for closed-shell references.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any singlet DIP states.
OPTIONS:
![$[i,j,k\ldots ]$](images/img-0890.png)
Find
RECOMMENDATION:
None
DIP_STATES
Sets the number of DIP roots to find. For closed-shell reference, defaults into DIP_SINGLETS. For open-shell references, specifies all low-lying states.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any DIP states.
OPTIONS:
Find
RECOMMENDATION:
None
DIP_TRIPLETS
Sets the number of triplet DIP roots to find. Valid only for closed-shell references.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any DIP triplet states.
OPTIONS:
Find
RECOMMENDATION:
None
DIRECT_SCF
Controls direct SCF.
TYPE:
LOGICAL
DEFAULT:
Determined by program.
OPTIONS:
TRUE
Forces direct SCF.
FALSE
Do not use direct SCF.
RECOMMENDATION:
Use the default; direct SCF switches off in-core integrals.
DISP_FREE_C
Specify the employed “dispersion-free" correlation functional.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
Correlation functionals supported by Q-Chem.
RECOMMENDATION:
Put the appropriate correlation functional paired with the chosen exchange functional (e.g. put PBE if DISP_FREE_X is revPBE); put NONE if DISP_FREE_X is set to an exchange-correlation functional.
DISP_FREE_X
Specify the employed “dispersion-free" exchange functional.
TYPE:
STRING
DEFAULT:
HF
OPTIONS:
Exchange functionals (e.g. revPBE) or exchange-correlation functionals (e.g. B3LYP)
supported by Q-Chem.
RECOMMENDATION:
HF is recommended for hybrid (primary) functionals (e.g.
B97X-V) and revPBE for semi-local ones (e.g.B97M-V). Other reasonable options (e.g. B3LYP for B3LYP-D3) can also be applied.
DORAMAN
Controls calculation of Raman intensities. Requires JOBTYPE to be set to FREQ
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not calculate Raman intensities.
TRUE
Do calculate Raman intensities.
RECOMMENDATION:
None
DSF_STATES
Sets the number of doubly spin-flipped target states roots to find.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any DSF states.
OPTIONS:
Find
RECOMMENDATION:
None
DUAL_BASIS_ENERGY
Activates dual-basis SCF (HF or DFT) energy correction.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Analytic first derivative available for HF and DFT (see JOBTYPE)
Can be used in conjunction with MP2 or RI-MP2
See BASIS, BASIS2, BASISPROJTYPE
RECOMMENDATION:
Use dual-basis to capture large-basis effects at smaller basis cost. Particularly useful with RI-MP2, in which HF often dominates. Use only proper subsets for small-basis calculation.
D_CPSCF_PERTNUM
Specifies whether to do the perturbations one at a time, or all together.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Perturbed densities to be calculated all together.
1
Perturbed densities to be calculated one at a time.
RECOMMENDATION:
None
D_SCF_CONV_1
Sets the convergence criterion for the level-1 iterations. This preconditions the density for the level-2 calculation, and does not include any two-electron integrals.
TYPE:
INTEGER
DEFAULT:
4
corresponding to a threshold of
.
OPTIONS:

Sets convergence threshold to
RECOMMENDATION:
The criterion for level-1 convergence must be less than or equal to the level-2 criterion, otherwise the D-CPSCF will not converge.
D_SCF_CONV_2
Sets the convergence criterion for the level-2 iterations.
TYPE:
INTEGER
DEFAULT:
4
Corresponding to a threshold of
OPTIONS:
Sets convergence threshold to
RECOMMENDATION:
None
D_SCF_DIIS
Specifies the number of matrices to use in the DIIS extrapolation in the D-CPSCF.
TYPE:
INTEGER
DEFAULT:
11
OPTIONS:
RECOMMENDATION:
Use the default.
D_SCF_MAX_1
Sets the maximum number of level-1 iterations.
TYPE:
INTEGER
DEFAULT:
100
OPTIONS:
User defined.
RECOMMENDATION:
Use the default.
D_SCF_MAX_2
Sets the maximum number of level-2 iterations.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
RECOMMENDATION:
Use the default.
EA_STATES
Sets the number of attached target states roots to find. By default,
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any EA states.
OPTIONS:
Find
RECOMMENDATION:
None
ECP
Defines the effective core potential and associated basis set to be used
TYPE:
STRING
DEFAULT:
No ECP
OPTIONS:
General, Gen
User defined. ($ecp keyword required)
Symbol
Use standard ECPs discussed above.
RECOMMENDATION:
ECPs are recommended for first row transition metals and heavier elements. Consul the reviews for more details.
EDA2
Switch on EDA2 and specify the option set number.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not run through EDA2.
1
Frozen energy decomposition + nDQ-FERF polarization
(the standard EDA2 option)
2
Frozen energy decomposition + (AO-block-based) ALMO polarization
(old scheme with the addition of frozen decomposition)
3
Frozen energy decomposition + oDQ-FERF polarization
(NOT commonly used)
4
Frozen wave function relaxation + Frozen energy decomposition + nDQ-FERF polarization
(NOT commonly used)
5
Frozen energy decomposition + polMO polarization
(NOT commonly used).
10
No preset. Completely controlled by user’s $rem input
(for developers only)
RECOMMENDATION:
Turn on EDA2 for Q-Chem’s ALMO-EDA jobs unless CTA with the old scheme is desired. Option 1 is recommended in general, especially when substantially large basis sets are employed. The original ALMO scheme (option 2) can be used when the employed basis set is of small or medium size (arguably no larger than augmented triple-
). The other options are rarely used for routine applications.
EDA_BSSE
Calculates the BSSE correction when performing the energy decomposition analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set to TRUE unless a very large basis set is used.
EDA_CLS_DISP
Compute the DISP contribution without performing the orthogonal decomposition, which will then be subtracted from the classical PAULI term.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Use the DISP term computed with orthogonal decomposition (if available).
TRUE
Use the DISP term computed using undistorted monomer densities.
RECOMMENDATION:
Set it to TRUE when orthogonal decomposition is not performed.
EDA_CLS_ELEC
Perform the classical decomposition of the frozen term.
TYPE:
BOOLEAN
DEFAULT:
FALSE (automatically set to TRUE by EDA2 options 1–5)
OPTIONS:
FALSE
Do not compute the classical ELEC and PAULI terms.
TRUE
Perform the classical decomposition.
RECOMMENDATION:
TRUE
EDA_COVP
Perform COVP analysis when evaluating the RS or ARS charge-transfer correction. COVP analysis is currently implemented only for systems of two fragments.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set to TRUE to perform COVP analysis in an EDA or SCF MI(RS) job.
EDA_PRINT_COVP
Replace the final MOs with the CVOP orbitals in the end of the run.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set to TRUE to print COVP orbitals instead of conventional MOs.
EE_SINGLETS
Controls the number of singlet excited states to calculate.
TYPE:
INTEGER/ARRAY
DEFAULT:
0
Do not perform an ADC calculation of singlet excited states
OPTIONS:
Number of singlet states to calculate for each irrep or
![$[n_1, n_2, ...]$](images/img-1109.png)
Compute
states for the first irrep,
states for the second irrep, ...
RECOMMENDATION:
Use this variable to define the number of excited states in case of restricted calculations of singlet states. In unrestricted calculations it can also be used, if EE_STATES not set. Then, it has the same effect as setting EE_STATES.
EE_STATES
Controls the number of excited states to calculate.
TYPE:
INTEGER/ARRAY
DEFAULT:
0
Do not perform an ADC calculation
OPTIONS:
Number of states to calculate for each irrep or
Compute
RECOMMENDATION:
Use this variable to define the number of excited states in case of unrestricted or open-shell calculations. In restricted calculations it can also be used, if neither EE_SINGLETS nor EE_TRIPLETS is given. Then, it has the same effect as setting EE_SINGLETS.
EE_TRIPLETS
Controls the number of triplet excited states to calculate.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not perform an ADC calculation of triplet excited states
OPTIONS:
Number of triplet states to calculate for each irrep or
Compute
RECOMMENDATION:
Use this variable to define the number of excited states in case of restricted calculations of triplet states.
EFP_COORD_XYZ
Use coordinates of three atoms instead of Euler angles to specify position and orientation of the fragments
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE FALSE
RECOMMENDATION:
None
EFP_DIRECT_POLARIZATION_DRIVER
Use direct solver for EFP polarization
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE FALSE
RECOMMENDATION:
Direct polarization solver provides stable convergence of induced dipoles which may otherwise become problematic in case of closely lying or highly polar or charged fragments. The computational cost of direct polarization versus iterative polarization becomes higher for systems containing more than 10000 polarizable points.
EFP_DISP_DAMP
Controls fragment-fragment dispersion screening in EFP
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
0
switch off dispersion screening
1
use Tang-Toennies screening, with fixed parameter

2
use overlap-based damping
RECOMMENDATION:
None
EFP_DISP
Controls fragment-fragment dispersion in EFP
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
switch on dispersion
FALSE
switch off dispersion
RECOMMENDATION:
None
EFP_ELEC_DAMP
Controls fragment-fragment electrostatic screening in EFP
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
0
switch off electrostatic screening
1
use overlap-based damping correction
2
use exponential damping correction if screening parameters are provided in the EFP potential
RECOMMENDATION:
Overlap-based damping is recommended
EFP_ELEC
Controls fragment-fragment electrostatics in EFP
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
switch on electrostatics
FALSE
switch off electrostatics
RECOMMENDATION:
None
EFP_ENABLE_LINKS
Enable fragment links in EFP region
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE FALSE
RECOMMENDATION:
None
EFP_EXREP
Controls fragment-fragment exchange repulsion in EFP
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
switch on exchange repulsion
FALSE
switch off exchange repulsion
RECOMMENDATION:
None
EFP_FRAGMENTS_ONLY
Specifies whether there is a QM part
TYPE:
LOGICAL
DEFAULT:
FALSE
QM part is present
OPTIONS:
TRUE
Only MM part is present: all fragments are treated by EFP
FALSE
QM part is present: do QM/MM EFP calculation
RECOMMENDATION:
None
EFP_INPUT
Specifies the format of EFP input
TYPE:
LOGICAL
DEFAULT:
FALSE
Dummy atom (e.g., He) in $molecule section should be present
OPTIONS:
TRUE
A format without dummy atom in $molecule section
FALSE
A format with dummy atom in $molecule section
RECOMMENDATION:
None
EFP_POL_DAMP
Controls fragment-fragment polarization screening in EFP
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
0
switch off polarization screening
1
use Tang-Toennies screening
RECOMMENDATION:
None
EFP_POL
Controls fragment-fragment polarization in EFP
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
switch on polarization
FALSE
switch off polarization
RECOMMENDATION:
None
EFP_QM_DISP
Controls QM-EFP dispersion
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
switch on QM-EFP dispersion
FALSE
switch off QM-EFP dispersion
RECOMMENDATION:
None
EFP_QM_ELEC_DAMP
Controls QM-EFP electrostatics screening in EFP
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
switch off electrostatic screening
1
use overlap based damping correction
RECOMMENDATION:
None
EFP_QM_ELEC
Controls QM-EFP electrostatics
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
switch on QM-EFP electrostatics
FALSE
switch off QM-EFP electrostatics
RECOMMENDATION:
None
EFP_QM_EXREP
Controls QM-EFP exchange-repulsion
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
switch on QM-EFP exchange-repulsion
FALSE
switch off QM-EFP exchange-repulsion
RECOMMENDATION:
None
EFP_QM_POL
Controls QM-EFP polarization
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
switch on QM-EFP polarization
FALSE
switch off QM-EFP polarization
RECOMMENDATION:
None
EFP
Specifies that EFP calculation is requested
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE FALSE
RECOMMENDATION:
The keyword should be present if excited state calculation is requested
EMBEDMAN
Turns density embedding on.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not use density embedding.
1
Turn on density embedding.
RECOMMENDATION:
Use EMBEDMAN for QM/QM density embedded calculations.
EMBED_MU
Specifies exponent value of projection operator scaling factor,
TYPE:
INTEGER
DEFAULT:
7
OPTIONS:
n
.
RECOMMENDATION:
Values of 2 - 7 are recommended. A higher value of
introduces numerical noise.
results in non-additive terms becoming too large. Energy corrections are fairly insensitive to changes in
.
EMBED_THEORY
Specifies post-DFT method performed on fragment one.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No post HF method, only DFT on fragment one.
1
Perform CCSD(T) calculation on fragment one.
2
Perform MP2 calculation on fragment one.
RECOMMENDATION:
This should be 1 or 2 for the high-level QM calculation of fragment 1-in-2, and 0 for fragment 2-in-1 low-level QM calculation.
EMBED_THRESH
Specifies threshold cutoff for AO contribution used to determine which MOs belong to which fragments
TYPE:
INTEGER
DEFAULT:
500
OPTIONS:
n
Threshold

RECOMMENDATION:
Acceptable values range from 0 to 1000. Should only need to be tuned for non-highly localized MOs
EOM_CORR
Specifies the correlation level.
TYPE:
STRING
DEFAULT:
None
No correction will be computed
OPTIONS:
SD(DT)
EOM-CCSD(dT), available for EE, SF, and IP
SD(FT)
EOM-CCSD(fT), available for EE, SF, IP, and EA
SD(ST)
EOM-CCSD(sT), available for IP
RECOMMENDATION:
None
EOM_DAVIDSON_CONVERGENCE
Convergence criterion for the RMS residuals of excited state vectors.
TYPE:
INTEGER
DEFAULT:
5
Corresponding to
OPTIONS:
Corresponding to
RECOMMENDATION:
Use the default. Normally this value be the same as EOM_DAVIDSON_THRESHOLD.
EOM_DAVIDSON_MAXVECTORS
Specifies maximum number of vectors in the subspace for the Davidson diagonalization.
TYPE:
INTEGER
DEFAULT:
60
OPTIONS:
Up to
RECOMMENDATION:
Larger values increase disk storage but accelerate and stabilize convergence.
EOM_DAVIDSON_MAX_ITER
Maximum number of iteration allowed for Davidson diagonalization procedure.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
User-defined number of iterations
RECOMMENDATION:
Default is usually sufficient
EOM_DAVIDSON_THRESHOLD
Specifies threshold for including a new expansion vector in the iterative Davidson diagonalization. Their norm must be above this threshold.
TYPE:
INTEGER
DEFAULT:
00103
Corresponding to 0.00001
OPTIONS:
Integer code is mapped to
, i.e., 02505->2.5
RECOMMENDATION:
Use the default unless converge problems are encountered. Should normally be set to the same values as EOM_DAVIDSON_CONVERGENCE, if convergence problems arise try setting to a value slightly larger than EOM_DAVIDSON_CONVERGENCE.
EOM_EA_ALPHA
Sets the number of attached target states derived by attaching
=
, default in EOM-EA).
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any EA states.
OPTIONS:
Find
RECOMMENDATION:
None
EOM_EA_BETA
Sets the number of attached target states derived by attaching
, EA-SF).
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any EA states.
OPTIONS:
Find
RECOMMENDATION:
None
EOM_FAKE_IPEA
If TRUE, calculates fake EOM-IP or EOM-EA energies and properties using the diffuse orbital trick. Default for EOM-EA and Dyson orbital calculations in CCMAN.
TYPE:
LOGICAL
DEFAULT:
FALSE (use proper EOM-IP code)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
None. This feature only works for CCMAN.
EOM_IPEA_FILTER
If TRUE, filters the EOM-IP/EA amplitudes obtained using the diffuse orbital implementation (see EOM_FAKE_IPEA). Helps with convergence.
TYPE:
LOGICAL
DEFAULT:
FALSE (EOM-IP or EOM-EA amplitudes will not be filtered)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
None
EOM_IP_ALPHA
Sets the number of ionized target states derived by removing
).
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any IP/
OPTIONS:
Find
RECOMMENDATION:
None
EOM_IP_BETA
Sets the number of ionized target states derived by removing
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any IP/
OPTIONS:
Find
RECOMMENDATION:
None
EOM_NGUESS_DOUBLES
Specifies number of excited state guess vectors which are double excitations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Include
RECOMMENDATION:
This should be set to the expected number of doubly excited states, otherwise they may not be found.
EOM_NGUESS_SINGLES
Specifies number of excited state guess vectors that are single excitations.
TYPE:
INTEGER
DEFAULT:
Equal to the number of excited states requested
OPTIONS:
Include
RECOMMENDATION:
Should be greater or equal than the number of excited states requested, unless .
EOM_POL
Whether or not the static polarizability for the EOM-CCSD wave function will be calculated.
TYPE:
LOGICAL
DEFAULT:
FALSE (EOM polarizability will not be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
Static polarizabilities are expensive since they require solving three additional response equations. Do no request this property unless you need it.
EOM_PRECONV_DOUBLES
When not zero, doubly excited vectors are converged prior to a full excited states calculation. Sets the maximum number of iterations for pre-converging procedure
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not pre-converge
N
Perform N Davidson iterations pre-converging doubles.
RECOMMENDATION:
Occasionally necessary to ensure a doubly excited state is found. Also used in DSF calculations instead of EOM_PRECONV_SINGLES
EOM_PRECONV_SD
When not zero, EOM vectors are pre-converged prior to a full excited states calculation. Sets the maximum number of iterations for pre-converging procedure.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
do not pre-converge
N
perform N Davidson iterations pre-converging singles and doubles.
RECOMMENDATION:
Occasionally necessary to ensure that all low-lying states are found. Also, very useful in EOM(2,3) calculations.
None
EOM_PRECONV_SINGLES
When not zero, singly excited vectors are converged prior to a full excited states calculation. Sets the maximum number of iterations for pre-converging procedure.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
do not pre-converge
1
pre-converge singles
RECOMMENDATION:
Sometimes helps with problematic convergence.
EOM_REF_PROP_TE
Request for calculation of non-relaxed two-particle EOM-CC properties. The two-particle properties currently include
TYPE:
LOGICAL
DEFAULT:
FALSE
(no two-particle properties will be calculated)
OPTIONS:
FALSE, TRUE
RECOMMENDATION:
The two-particle properties are computationally expensive since they require calculation and use of the two-particle density matrix (the cost is approximately the same as the cost of an analytic gradient calculation). Do not request the two-particle properties unless you really need them.
EOM_SHIFT
Specifies energy shift in EOM calculations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
corresponds to
hartree shift (i.e., 11000 = 11 hartree); solve for eigenstates around this value.
RECOMMENDATION:
Not available in CCMAN.
EOM_USER_GUESS
Specifies if user-defined guess will be used in EOM calculations.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Solve for a state that has maximum overlap with a trans-n specified in $eom_user_guess.
RECOMMENDATION:
The orbitals are ordered by energy, as printed in the beginning of the CCMAN2 output. Not available in CCMAN.
EPAO_ITERATE
Controls iterations for EPAO calculations (see PAO_METHOD).
TYPE:
INTEGER
DEFAULT:
0
Use non-iterated EPAOs based on atomic blocks of SPS.
OPTIONS:
Optimize the EPAOs for up to
RECOMMENDATION:
Use the default. For molecules that are not too large, one can test the sensitivity of the results to the type of minimal functions by the use of optimized EPAOs in which case a value of
is reasonable.
EPAO_WEIGHTS
Controls algorithm and weights for EPAO calculations (see PAO_METHOD).
TYPE:
INTEGER
DEFAULT:
115
Standard weights, use 1
and 2
OPTIONS:
15
Standard weights, with 1
RECOMMENDATION:
Use the default, unless convergence failure is encountered.
ERCALC
Specifies the Edmiston-Ruedenberg localized orbitals are to be calculated
TYPE:
INTEGER
DEFAULT:
06000
OPTIONS:


specifies the convergence threshold.
If
, the threshold is set to
. The default is 6.
If
, the calculation is aborted after the guess, allowing Pipek-Mezey
orbitals to be extracted.

specifies the guess:
0 Boys localized orbitals. This is the default
1 Pipek-Mezey localized orbitals.
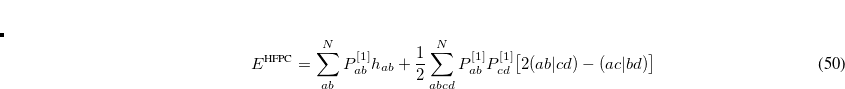
specifies restart options (if restarting from an ER calculation):
0 No restart. This is the default
1 Read in MOs from last ER calculation.
2 Read in MOs and RI integrals from last ER calculation.

specifies how to treat core orbitals
0 Do not perform ER localization. This is the default.
1 Localize core and valence together.
2 Do separate localizations on core and valence.
3 Localize only the valence electrons.
4 Use the $localize section.
RECOMMENDATION:
ERCALC 1 will usually suffice, which uses threshold
ER_CIS_NUMSTATE
Define how many states to mix with ER localized diabatization. These states must be specified in the $localized_diabatization section.
TYPE:
INTEGER
DEFAULT:
0
Do not perform ER localized diabatization.
OPTIONS:
2 to N where N is the number of CIS states requested (CIS_N_ROOTS)
RECOMMENDATION:
It is usually not wise to mix adiabatic states that are separated by more than a few eV or a typical reorganization energy in solvent.
ESP_TRANS
Controls the calculation of the electrostatic potential of the transition density
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
compute the electrostatic potential of the excited state transition density
FALSE
compute the electrostatic potential of the excited state electronic density
RECOMMENDATION:
NONE
EXCHANGE
Specifies the exchange functional (or most exchange-correlation functionals for backwards compatibility).
TYPE:
STRING
DEFAULT:
No default
OPTIONS:
NAME
Use EXCHANGE = NAME, where NAME is either:
1) One of the exchange functionals listed in Section 5.3.2
2) One of the XC functionals listed in Section 5.3.4 that is not marked with an
asterisk.
3) GEN, for a user-defined functional (see Section 5.3.6).
RECOMMENDATION:
In general, consult the literature to guide your selection. Our recommendations are indicated in bold in Sections 5.3.4 and 5.3.2.
FAST_XC
Controls direct variable thresholds to accelerate exchange-correlation (XC) in DFT.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Turn FAST_XC on.
FALSE
Do not use FAST_XC.
RECOMMENDATION:
Caution: FAST_XC improves the speed of a DFT calculation, but may occasionally cause the SCF calculation to diverge.
FDE
Turns density embedding on.
TYPE:
BOOLEAN
DEFAULT:
False
OPTIONS:
True
Perform an FDE-ADC calculation.
False
Don’t perform FDE-ADC calculation.
RECOMMENDATION:
Set the $rem variable FDE to TRUE to start a FDE-ADC calculation.
FDIFF_DER
Controls what types of information are used to compute higher derivatives. The default uses a combination of energy, gradient and Hessian information, which makes the force field calculation faster.
TYPE:
INTEGER
DEFAULT:
3
for jobs where analytical 2nd derivatives are available.
0
for jobs with ECP.
OPTIONS:
0
Use energy information only.
1
Use gradient information only.
2
Use Hessian information only.
3
Use energy, gradient, and Hessian information.
RECOMMENDATION:
When the molecule is larger than benzene with small basis set, FDIFF_DER=2 may be faster. Note that FDIFF_DER will be set lower if analytic derivatives of the requested order are not available. Please refers to IDERIV.
FDIFF_STEPSIZE_QFF
Displacement used for calculating third and fourth derivatives by finite difference.
TYPE:
INTEGER
DEFAULT:
5291
Corresponding to 0.1 bohr. For calculating third and fourth derivatives.
OPTIONS:
Use a step size of
.
RECOMMENDATION:
Use the default, unless the potential surface is very flat, in which case a larger value should be used.
FDIFF_STEPSIZE
Displacement used for calculating derivatives by finite difference.
TYPE:
INTEGER
DEFAULT:
100
Corresponding to 0.001 Å. For calculating second derivatives.
OPTIONS:
Use a step size of
RECOMMENDATION:
Use the default except in cases where the potential surface is very flat, in which case a larger value should be used. See FDIFF_STEPSIZE_QFF for third and fourth derivatives.
FD_MAT_VEC_PROD
Compute Hessian-vector product using the finite difference technique. |
TYPE:
BOOLEAN |
DEFAULT:
FALSE (TRUE when the employed functional contains NLC) |
OPTIONS:
FALSE |
Compute Hessian-vector product analytically. |
TRUE |
Use finite difference to compute Hessian-vector product. |
RECOMMENDATION:
Set it to TRUE when analytical Hessian is not available. |
Note: |
only the NLC part will be computed with finite difference.
FEFP_EFP
Specifies that fEFP_EFP calculation is requested to compute the total interaction energies between a ligand (the last fragment in the $efp_fragments section) and the protein (represented by fEFP)
TYPE:
STRING
DEFAULT:
OFF
OPTIONS:
OFF
disables fEFP
LA
enables fEFP with the Link Atom (HLA or CLA) scheme (only electrostatics and polarization)
MFCC
enables fEFP with MFCC (only electrostatics)
RECOMMENDATION:
The keyword should be invoked if EFP/fEFP is requested (interaction energy calculations). This keyword has to be employed with EFP_FRAGMENT_ONLY = TRUE. To switch on/off electrostatics or polarzation interactions, the usual EFP controls are employed.
FEFP_QM
Specifies that fEFP_QM calculation is requested to perform a QM/fEFPcompute computation. The fEFP part is a fractionated macromolecule.
TYPE:
STRING
DEFAULT:
OFF
OPTIONS:
OFF
disables fEFP_QM and performs a QM/EFP calculation
LA
enables fEFP_QM with the Link Atom scheme
RECOMMENDATION:
The keyword should be invoked if QM/fEFP is requested. This keyword has to be employed with efp_fragment_only false. Only electrostatics is available.
FMO_ORDER
Controls the truncation order
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
Order of FMO
RECOMMENDATION:
FMO can be performed up to third order.
FOA_FUNDGAP
Compute the frozen-orbital approximation of the fundamental gap.
TYPE:
Boolean
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not compute FOA derivative discontinuity and fundamental gap.
TRUE
Compute and print FOA fundamental gap information. Implies KS_GAP_PRINT.
RECOMMENDATION:
Use in conjunction with KS_GAP_UNIT if true.
FOCK_EXTRAP_ORDER
Specifies the polynomial order
TYPE:
INTEGER
DEFAULT:
0
Do not perform Fock matrix extrapolation.
OPTIONS:
Extrapolate using an
).
RECOMMENDATION:
None
FOCK_EXTRAP_POINTS
Specifies the number
of old Fock matrices that are retained for use in extrapolation.
TYPE:
INTEGER
DEFAULT:
0
Do not perform Fock matrix extrapolation.
OPTIONS:
Save

RECOMMENDATION:
Higher-order extrapolations with more saved Fock matrices are faster and conserve energy better than low-order extrapolations, up to a point. In many cases, the scheme (
FOLLOW_ENERGY
Adjusts the energy window for near states
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use dynamic thresholds, based on energy difference between steps.
Search over selected state
.
RECOMMENDATION:
Use a wider energy window to follow a state diabatically, smaller window to remain on the adiabatic state most of the time.
FOLLOW_OVERLAP
Adjusts the threshold for states of similar character.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use dynamic thresholds, based on energy difference between steps.
Percentage overlap for previous step and current step.
RECOMMENDATION:
Use a higher value to require states have higher degree of similarity to be considered the same (more often selected based on energy).
FON_E_THRESH
DIIS error below which occupations will be kept constant.
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
n
freeze occupations below DIIS error of
RECOMMENDATION:
This should be one or two numbers bigger than the desired SCF convergence threshold.
FON_NORB
Number of orbitals above and below the Fermi level that are allowed to have fractional occupancies.
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
n
number of active orbitals
RECOMMENDATION:
The number of valence orbitals is a reasonable choice.
FON_T_END
Final electronic temperature for FON calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Any desired final temperature.
RECOMMENDATION:
Pick the temperature to either reproduce experimental conditions (e.g. room temperature) or as low as possible to approach zero-temperature.
FON_T_METHOD
Selects cooling algorithm.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
temperature is scaled by a factor in each cycle
2
temperature is decreased by a constant number in each cycle
RECOMMENDATION:
We have made slightly better experience with a constant cooling rate. However, choose constant temperature when in doubt.
FON_T_SCALE
Determines the step size for the cooling.
TYPE:
INTEGER
DEFAULT:
90
OPTIONS:
n
temperature is scaled by
in each cycle (cooling method 1)
n
temperature is decreased by n K in each cycle (cooling method 2)
RECOMMENDATION:
The cooling rate should be neither too slow nor too fast. Too slow may lead to final energies that are at undesirably high temperatures. Too fast may lead to convergence issues. Reasonable choices for methods 1 and 2 are 98 and 50, respectively. When in doubt, use constant temperature.
FON_T_START
Initial electronic temperature (in K) for FON calculation.
TYPE:
INTEGER
DEFAULT:
1000
OPTIONS:
Any desired initial temperature.
RECOMMENDATION:
Pick the temperature to either reproduce experimental conditions (e.g. room temperature) or as low as possible to approach zero-temperature.
FORCE_FIELD
Specifies the force field for MM energies in QM/MM calculations.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
AMBER99
AMBER99 force field
CHARMM27
CHARMM27 force field
OPLSAA
OPLSAA force field
RECOMMENDATION:
None.
FRACTIONAL_ELECTRON
Add or subtract a fraction of an electron.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use an integer number of electrons.
Add
electrons to the system.
RECOMMENDATION:
Use only if trying to generate
plots. If
, a fraction of an electron is removed from the system.
FRAGMO_GUESS_MODE
Decide what to do regarding to the FRAGMO guess in the present job.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Spawn fragment jobs sequentially and collect the results as the FRAGMO guess at the end.
1
Generate fragment inputs in folders “FrgX" under the scratch directory of the present job
and then terminate. Users can then take advantage of a queuing system to run these jobs
simultaneously using “FrgX" as their scratch folders (should be handled with scripting).
2
Read in the available fragment data.
RECOMMENDATION:
Consider using “1" if the fragment calculations are evenly expensive. Use “2" when FRAGMO guess is pre-computed.
FRAG_MOL_ORB
Perform a FMO calculation.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform a FMO calculation.
FALSE
Do not perform a FMO calculation.
RECOMMENDATION:
NONE
FRGM_LPCORR
Specifies a correction method performed after the locally-projected equations are converged.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
ARS
Approximate Roothaan-step perturbative correction.
RS
Single Roothaan-step perturbative correction.
EXACT_SCF
Full SCF variational correction.
ARS_EXACT_SCF
Both ARS and EXACT_SCF in a single job.
RS_EXACT_SCF
Both RS and EXACT_SCF in a single job.
RECOMMENDATION:
For large basis sets use ARS, use RS if ARS fails.
FRGM_METHOD
Specifies a locally-projected method.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
STOLL
Locally-projected SCF equations of Stoll are solved.
GIA
Locally-projected SCF equations of Gianinetti are solved.
NOSCF_RS
Single Roothaan-step correction to the FRAGMO initial guess.
NOSCF_ARS
Approximate single Roothaan-step correction to the FRAGMO initial guess.
NOSCF_DRS
Double Roothaan-step correction to the FRAGMO initial guess.
NOSCF_RS_FOCK
Non-converged SCF energy of the single Roothaan-step MOs.
RECOMMENDATION:
STOLL and GIA are for variational optimization of the ALMOs. NOSCF options are for computationally fast corrections of the FRAGMO initial guess.
FRZ_GEOM
Compute forces on the frozen PES.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not compute forces on the frozen PES.
TRUE
Compute forces on the frozen PES.
RECOMMENDATION:
Set it to TRUE when optimized geometry or vibrational frequencies on the frozen PES are desired.
FRZ_ORTHO_DECOMP_CONV
Convergence criterion for the minimization problem that gives the orthogonal fragment densities.
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
RECOMMENDATION:
Use the default unless tighter convergence is preferred.
FRZ_ORTHO_DECOMP
Perform the decomposition of frozen interaction energy based on the orthogonal decomposition of the 1PDM associated with the frozen wave function.
TYPE:
BOOLEAN
DEFAULT:
FALSE (automatically set to TRUE by EDA2 options 1–5)
OPTIONS:
FALSE
Do not perform the orthogonal decomposition.
TRUE
Perform the frozen energy decomposition using orthogonal fragment densities.
RECOMMENDATION:
Use default value automatically set by “EDA2".
FSM_MODE
Specifies the method of interpolation
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
1
Cartesian
2
LST
RECOMMENDATION:
In most cases, LST is superior to Cartesian interpolation.
FSM_NGRAD
Specifies the number of perpendicular gradient steps used to optimize each node
TYPE:
INTEGER
DEFAULT:
Undefined
OPTIONS:
Number of perpendicular gradients per node
RECOMMENDATION:
Anything between 2 and 6 should work, where increasing the number is only needed for difficult reaction paths.
FSM_NNODE
Specifies the number of nodes along the string
TYPE:
INTEGER
DEFAULT:
Undefined
OPTIONS:
number of nodes in FSM calculation
RECOMMENDATION:
. Use 10 to 20 nodes for a typical calculation. Reaction paths that connect multiple elementary steps should be separated into individual elementary steps, and one FSM job run for each pair of intermediates. Use a higher number when the FSM is followed by an approximate-Hessian based transition state search (Section 10.2.2).
FSM_OPT_MODE
Specifies the method of optimization
TYPE:
INTEGER
DEFAULT:
Undefined
OPTIONS:
1
Conjugate gradients
2
Quasi-Newton method with BFGS Hessian update
RECOMMENDATION:
The quasi-Newton method is more efficient when the number of nodes is high.
FSSH_CONTINUE
Restart a FSSH calculation from a previous run, using the file 396.0. When this is enabled, the initial conditions of the surface hopping calculation will be set, including the correct wave function amplitudes, initial surface, and position/momentum moments (if AFSSH) from the final step of some prior calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Start fresh calculation.
Restart from previous run.
RECOMMENDATION:
None
FSSH_INITIALSURFACE
Specifies the initial state in a surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
An integer between FSSH_LOWESTSURFACE and FSSH_LOWESTSURFACE
FSSH_NSURFACES
.
RECOMMENDATION:
None
FSSH_LOWESTSURFACE
Specifies the lowest-energy state considered in a surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
Only states
RECOMMENDATION:
None
FSSH_NSURFACES
Specifies the number of states considered in a surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
RECOMMENDATION:
Any states which may come close in energy to the active surface should be included in the surface hopping calculation.
FTC_CLASS_THRESH_MULT
Together with FTC_CLASS_THRESH_ORDER, determines the cutoff threshold for included a shell-pair in the
class, i.e., the class that is expanded in terms of plane waves.
TYPE:
INTEGER
DEFAULT:
5
Multiplicative part of the FTC classification threshold. Together with
the default value of the FTC_CLASS_THRESH_ORDER this leads to
the
threshold value.
OPTIONS:
User specified.
RECOMMENDATION:
Use the default. If diffuse basis sets are used and the molecule is relatively big then tighter FTC classification threshold has to be used. According to our experiments using Pople-type diffuse basis sets, the default
threshold value for alanine10 and
value for alanine15 has to be used.
FTC_CLASS_THRESH_ORDER
Together with FTC_CLASS_THRESH_MULT, determines the cutoff threshold for included a shell-pair in the
TYPE:
INTEGER
DEFAULT:
5
Logarithmic part of the FTC classification threshold. Corresponds to
OPTIONS:
User specified
RECOMMENDATION:
Use the default.
FTC_SMALLMOL
Controls whether or not the operator is evaluated on a large grid and stored in memory to speed up the calculation.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Use a big pre-calculated array to speed up the FTC calculations
0
Use this option to save some memory
RECOMMENDATION:
Use the default if possible and use 0 (or buy some more memory) when needed.
FTC
Controls the overall use of the FTC.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not use FTC in the Coulomb part
1
Use FTC in the Coulomb part
RECOMMENDATION:
Use FTC when bigger and/or diffuse basis sets are used.
GAUSSIAN_BLUR
Enables the use of Gaussian-delocalized external charges in a QM/MM calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Delocalizes external charges with Gaussian functions.
FALSE
Point charges
RECOMMENDATION:
None
GAUSS_BLUR_WIDTH
Delocalization width for external MM Gaussian charges in a Janus calculations.
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
Use a width of
Å.
RECOMMENDATION:
Blur all MM external charges in a QM/MM calculation with the specified width. Gaussian blurring is currently incompatible with PCM calculations. Values of 1.0–2.0 Å are recommended in Ref. Das:2002.
GEN_SCFMAN_ALGO_1
The first algorithm to be used in a hybrid-algorithm calculation.
TYPE:
STRING
DEFAULT:
0
OPTIONS:
All the available SCF_ALGORITHM options, including the GEN_SCFMAN additions (Section 4.3.1).
RECOMMENDATION:
None
GEN_SCFMAN_CONV_1
The convergence criterion given to the first algorithm. If reached, switch to the next algorithm.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
10
RECOMMENDATION:
None
GEN_SCFMAN_HYBRID_ALGO
Use multiple algorithms in an SCF calculation based on GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Use a single SCF algorithm (given by SCF_ALGORITHM).
TRUE
Use multiple SCF algorithms (to be specified).
RECOMMENDATION:
Set it to TRUE when the use of more than one algorithm is desired.
GEN_SCFMAN_ITER_1
Maximum number of iterations given to the first algorithm. If used up, switch to the next algorithm.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
User-defined
RECOMMENDATION:
None
GEN_SCFMAN
Use GEN_SCFMAN for the present SCF calculation.
TYPE:
BOOLEAN
DEFAULT:
TRUE
OPTIONS:
FALSE
Use the previous SCF code.
TRUE
Use GEN_SCFMAN.
RECOMMENDATION:
Set to FALSE in cases where features not yet supported by GEN_SCFMAN are needed.
GEOM_OPT_CHARAC_CONV
Overide the built-in convergence criterion for the Davidson solver.
TYPE:
INTEGER
DEFAULT:
0 (use the built-in default value 10
)
OPTIONS:
Set the convergence criterion to 10
RECOMMENDATION:
Use the default. If it fails to converge, consider loosening the criterion with caution.
GEOM_OPT_CHARAC
Use the finite difference Davidson method to characterize the resulting energy minimum/transition state.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
do not characterize the resulting stationary point.
TRUE
perform a characterization of the stationary point.
RECOMMENDATION:
Set it to TRUE when the character of a stationary point needs to be verified, especially for a transition structure.
GEOM_OPT_COORDS
Controls the type of optimization coordinates.
TYPE:
INTEGER
DEFAULT:
OPTIONS:
0
Optimize in Cartesian coordinates.
1
Generate and optimize in internal coordinates, if this fails abort.
Generate and optimize in internal coordinates, if this fails at any stage of the
optimization, switch to Cartesian and continue.
2
Optimize in
Optimize in
optimization switch to Cartesians and continue.
RECOMMENDATION:
Use the default; delocalized internals are more efficient.
GEOM_OPT_DMAX
Maximum allowed step size. Value supplied is multiplied by 10
.
TYPE:
INTEGER
DEFAULT:
300
= 0.3
OPTIONS:
User-defined cutoff.
RECOMMENDATION:
Use the default.
GEOM_OPT_HESSIAN
Determines the initial Hessian status.
TYPE:
STRING
DEFAULT:
DIAGONAL
OPTIONS:
DIAGONAL
Set up diagonal Hessian.
READ
Have exact or initial Hessian. Use as is if Cartesian, or transform
if internals.
RECOMMENDATION:
An accurate initial Hessian will improve the performance of the optimizer, but is expensive to compute.
GEOM_OPT_LINEAR_ANGLE
Threshold for near linear bond angles (degrees).
TYPE:
INTEGER
DEFAULT:
165 degrees.
OPTIONS:
User-defined level.
RECOMMENDATION:
Use the default.
GEOM_OPT_MAX_CYCLES
Maximum number of optimization cycles.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
User defined positive integer.
RECOMMENDATION:
The default should be sufficient for most cases. Increase if the initial guess geometry is poor, or for systems with shallow potential wells.
GEOM_OPT_MAX_DIIS
Controls maximum size of subspace for GDIIS.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not use GDIIS.
-1
Default size = min(NDEG, NATOMS, 4) NDEG = number of molecular
degrees of freedom.
Size specified by user.
RECOMMENDATION:
Use the default or do not set
GEOM_OPT_MODE
Determines Hessian mode followed during a transition state search.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Mode following off.
Maximize along mode
RECOMMENDATION:
Use the default, for geometry optimizations.
GEOM_OPT_PRINT
Controls the amount of Optimize print output.
TYPE:
INTEGER
DEFAULT:
3
Error messages, summary, warning, standard information and gradient print out.
OPTIONS:
0
Error messages only.
1
Level 0 plus summary and warning print out.
2
Level 1 plus standard information.
3
Level 2 plus gradient print out.
4
Level 3 plus Hessian print out.
5
Level 4 plus iterative print out.
6
Level 5 plus internal generation print out.
7
Debug print out.
RECOMMENDATION:
Use the default.
GEOM_OPT_SYMFLAG
Controls the use of symmetry in Optimize.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Make use of point group symmetry.
FALSE
Do not make use of point group symmetry.
RECOMMENDATION:
Use the default.
GEOM_OPT_TOL_DISPLACEMENT
Convergence on maximum atomic displacement.
TYPE:
INTEGER
DEFAULT:
1200
tolerance on maximum atomic displacement.
OPTIONS:
Integer value (tolerance =
).
RECOMMENDATION:
Use the default. To converge GEOM_OPT_TOL_GRADIENT and one of GEOM_OPT_TOL_DISPLACEMENT and GEOM_OPT_TOL_ENERGY must be satisfied.
GEOM_OPT_TOL_ENERGY
Convergence on energy change of successive optimization cycles.
TYPE:
INTEGER
DEFAULT:
100
tolerance on maximum (absolute) energy change.
OPTIONS:
).
RECOMMENDATION:
Use the default. To converge GEOM_OPT_TOL_GRADIENT and one of GEOM_OPT_TOL_DISPLACEMENT and GEOM_OPT_TOL_ENERGY must be satisfied.
GEOM_OPT_TOL_GRADIENT
Convergence on maximum gradient component.
TYPE:
INTEGER
DEFAULT:
300
tolerance on maximum gradient component.
OPTIONS:
Integer value (tolerance =
RECOMMENDATION:
Use the default. To converge GEOM_OPT_TOL_GRADIENT and one of GEOM_OPT_TOL_DISPLACEMENT and GEOM_OPT_TOL_ENERGY must be satisfied.
GEOM_OPT_UPDATE
Controls the Hessian update algorithm.
TYPE:
INTEGER
DEFAULT:
-1
OPTIONS:
-1
Use the default update algorithm.
0
Do not update the Hessian (not recommended).
1
Murtagh-Sargent update.
2
Powell update.
3
Powell/Murtagh-Sargent update (TS default).
4
BFGS update (OPT default).
5
BFGS with safeguards to ensure retention of positive definiteness
(GDISS default).
RECOMMENDATION:
Use the default.
GEOM_PRINT
Controls the amount of geometric information printed at each step.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Prints out all geometric information; bond distances, angles, torsions.
FALSE
Normal printing of distance matrix.
RECOMMENDATION:
Use if you want to be able to quickly examine geometric parameters at the beginning and end of optimizations. Only prints in the beginning of single point energy calculations.
GHF
Run a generalized Hartree-Fock calculation with GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Run a GHF calculation.
FALSE
Do not use GHF.
RECOMMENDATION:
Set to TRUE if desired.
GRAIN
Controls the number of lowest-level boxes in one dimension for CFMM.
TYPE:
INTEGER
DEFAULT:
-1
Program decides best value, turning on CFMM when useful
OPTIONS:
-1
Program decides best value, turning on CFMM when useful
1
Do not use CFMM
Use CFMM with
RECOMMENDATION:
This is an expert option; either use the default, or use a value of 1 if CFMM is not desired.
GVB_AMP_SCALE
Scales the default orbital amplitude iteration step size by
TYPE:
INTEGER
DEFAULT:
1000
Corresponding to 100%
OPTIONS:
User-defined, 0–1000
RECOMMENDATION:
Default is usually fine, but in some highly-correlated systems it can help with convergence to use smaller values.
GVB_DO_ROHF
Sets the number of Unrestricted-in-Active Pairs to be kept restricted.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-Defined
RECOMMENDATION:
If
GVB_DO_SANO
Sets the scheme used in determining the active virtual orbitals in a Unrestricted-in-Active Pairs GVB calculation.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
0
No localization or Sano procedure
1
Only localizes the active virtual orbitals
2
Uses the Sano procedure
RECOMMENDATION:
Different initial guesses can sometimes lead to different solutions. Disabling sometimes can aid in finding more non-local solutions for the orbitals.
GVB_GUESS_MIX
Similar to SCF_GUESS_MIX, it breaks alpha/beta symmetry for UPP by mixing the alpha HOMO and LUMO orbitals according to the user-defined fraction of LUMO to add the HOMO. 100 corresponds to a 1:1 ratio of HOMO and LUMO in the mixed orbitals.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined,
RECOMMENDATION:
25 often works well to break symmetry without overly impeding convergence.
GVB_LOCAL
Sets the localization scheme used in the initial guess wave function.
TYPE:
INTEGER
DEFAULT:
2
Pipek-Mezey orbitals
OPTIONS:
0
No Localization
1
Boys localized orbitals
2
Pipek-Mezey orbitals
RECOMMENDATION:
Different initial guesses can sometimes lead to different solutions. It can be helpful to try both to ensure the global minimum has been found.
GVB_N_PAIRS
Alternative to CC_REST_OCC and CC_REST_VIR for setting active space size in GVB and valence coupled cluster methods.
TYPE:
INTEGER
DEFAULT:
PP active space (1 occ and 1 virt for each valence electron pair)
OPTIONS:
user-defined
RECOMMENDATION:
Use the default unless one wants to study a special active space. When using small active spaces, it is important to ensure that the proper orbitals are incorporated in the active space. If not, use the $reorder_mo feature to adjust the SCF orbitals appropriately.
GVB_OLD_UPP
Which unrestricted algorithm to use for GVB.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use Unrestricted-in-Active Pairs described in Ref. Lawler:2010
1
Use Unrestricted Implementation described in Ref. Beran:2005
RECOMMENDATION:
Only works for Unrestricted PP and no other GVB model.
GVB_ORB_CONV
The GVB-CC wave function is considered converged when the root-mean-square orbital gradient and orbital step sizes are less than
. Adjust THRESH simultaneously.
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
User-defined
RECOMMENDATION:
Use 6 for PP(2) jobs or geometry optimizations. Tighter convergence (i.e. 7 or higher) cannot always be reliably achieved.
GVB_ORB_MAX_ITER
Controls the number of orbital iterations allowed in GVB-CC calculations. Some jobs, particularly unrestricted PP jobs can require 500–1000 iterations.
TYPE:
INTEGER
DEFAULT:
256
OPTIONS:
User-defined number of iterations.
RECOMMENDATION:
Default is typically adequate, but some jobs, particularly UPP jobs, can require 500–1000 iterations if converged tightly.
GVB_ORB_SCALE
Scales the default orbital step size by
TYPE:
INTEGER
DEFAULT:
1000
Corresponding to 100%
OPTIONS:
User-defined, 0–1000
RECOMMENDATION:
Default is usually fine, but for some stretched geometries it can help with convergence to use smaller values.
GVB_POWER
Coefficient for GVB_IP exchange type amplitude regularization to improve the convergence of the amplitude equations especially for spin-unrestricted amplitudes near dissociation. This is the leading coefficient for an amplitude dampening term included in the energy denominator: -(
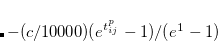
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:

User-defined
RECOMMENDATION:
Should be decreased if unrestricted amplitudes do not converge or converge slowly at dissociation, and should be kept even valued.
GVB_PRINT
Controls the amount of information printed during a GVB-CC job.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined
RECOMMENDATION:
Should never need to go above 0 or 1.
GVB_REGULARIZE
Coefficient for GVB_IP exchange type amplitude regularization to improve the convergence of the amplitude equations especially for spin-unrestricted amplitudes near dissociation. This is the leading coefficient for an amplitude dampening term
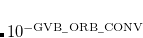
TYPE:
INTEGER
DEFAULT:
0
For restricted
1
For unrestricted
OPTIONS:
User-defined
RECOMMENDATION:
Should be increased if unrestricted amplitudes do not converge or converge slowly at dissociation. Set this to zero to remove all dynamically-valued amplitude regularization.
GVB_REORDER_1
Tells the code which two pairs to swap first.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined XXXYYY
RECOMMENDATION:
This is in the format of two 3-digit pair indices that tell the code to swap pair XXX with YYY, for example swapping pair 1 and 2 would get the input 001002. Must be specified in GVB_REORDER_PAIRS
1.
GVB_REORDER_2
Tells the code which two pairs to swap second.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined XXXYYY
RECOMMENDATION:
This is in the format of two 3-digit pair indices that tell the code to swap pair XXX with YYY, for example swapping pair 1 and 2 would get the input 001002. Must be specified in GVB_REORDER_PAIRS
GVB_REORDER_3
Tells the code which two pairs to swap third.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined XXXYYY
RECOMMENDATION:
This is in the format of two 3-digit pair indices that tell the code to swap pair XXX with YYY, for example swapping pair 1 and 2 would get the input 001002. Must be specified in GVB_REORDER_PAIRS
GVB_REORDER_4
Tells the code which two pairs to swap fourth.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined XXXYYY
RECOMMENDATION:
This is in the format of two 3-digit pair indices that tell the code to swap pair XXX with YYY, for example swapping pair 1 and 2 would get the input 001002. Must be specified in GVB_REORDER_PAIRS
GVB_REORDER_5
Tells the code which two pairs to swap fifth.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined XXXYYY
RECOMMENDATION:
This is in the format of two 3-digit pair indices that tell the code to swap pair XXX with YYY, for example swapping pair 1 and 2 would get the input 001002. Must be specified in GVB_REORDER_PAIRS
GVB_REORDER_PAIRS
Tells the code how many GVB pairs to switch around.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
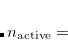
RECOMMENDATION:
This allows for the user to change the order the active pairs are placed in after the orbitals are read in or are guessed using localization and the Sano procedure. Up to 5 sequential pair swaps can be made, but it is best to leave this alone.
GVB_RESTART
Restart a job from previously-converged GVB-CC orbitals.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Useful when trying to converge to the same GVB solution at slightly different geometries, for example.
GVB_SHIFT
Value for a statically valued energy shift in the energy denominator used to solve the coupled cluster amplitude equations,
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined
RECOMMENDATION:
Default is fine, can be used in lieu of the dynamically valued amplitude regularization if it does not aid convergence.
GVB_SYMFIX
Should GVB use a symmetry breaking fix.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
no symmetry breaking fix
1
symmetry breaking fix with virtual orbitals spanning the active space
2
symmetry breaking fix with virtual orbitals spanning the whole virtual space
RECOMMENDATION:
It is best to stick with type 1 to get a symmetry breaking correction with the best results coming from CORRELATION=NP and GVB_SYMFIX = 1.
GVB_SYMPEN
Sets the pre-factor for the amplitude regularization term for the SB amplitudes.
TYPE:
INTEGER
DEFAULT:
160
OPTIONS:

User-defined
RECOMMENDATION:
Sets the pre-factor for the amplitude regularization term for the SB amplitudes:
.
GVB_SYMSCA
Sets the weight for the amplitude regularization term for the SB amplitudes.
TYPE:
INTEGER
DEFAULT:
125
OPTIONS:
User-defined
RECOMMENDATION:
Sets the weight for the amplitude regularization term for the SB amplitudes:
GVB_TRUNC_OCC
Controls how many pairs’ occupied orbitals are truncated from the GVB active space.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined
RECOMMENDATION:
This allows for asymmetric GVB active spaces removing the
GVB_TRUNC_VIR
Controls how many pairs’ virtual orbitals are truncated from the GVB active space.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined
RECOMMENDATION:
This allows for asymmetric GVB active spaces removing the
GVB_UNRESTRICTED
Controls restricted versus unrestricted PP jobs. Usually handled automatically.
TYPE:
LOGICAL
DEFAULT:
same value as UNRESTRICTED
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set this variable explicitly only to do a UPP job from an RHF or ROHF initial guess. Leave this variable alone and specify UNRESTRICTED = TRUE to access the new Unrestricted-in-Active-Pairs GVB code which can return an RHF or ROHF solution if used with GVB_DO_ROHF
HESS_AND_GRAD
Enables the evaluation of both analytical gradient and Hessian in a single job
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Evaluates both gradient and Hessian.
FALSE
Evaluates Hessian only.
RECOMMENDATION:
Use only in a frequency (and thus Hessian) evaluation.
HFPT_BASIS
Specifies the secondary basis in a HFPC/DFPC calculation.
TYPE:
STRING
DEFAULT:
None
OPTIONS:
None
RECOMMENDATION:
See reference for recommended basis set, functional, and grid pairings.
HFPT
Activates HFPC/DFPC calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Single-point energy only
RECOMMENDATION:
Use Dual-Basis to capture large-basis effects at smaller basis cost. See reference for recommended basis set, functional, and grid pairings.
HF_LR
Sets the fraction of Hartree-Fock exchange at
.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to HF_LR =
RECOMMENDATION:
None
HF_SR
Sets the fraction of Hartree-Fock exchange at
.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to HF_SR =
RECOMMENDATION:
None
HIRSHFELD_CONV
Set different SCF convergence criterion for the calculation of the single-atom Hirshfeld calculations
TYPE:
INTEGER
DEFAULT:
same as SCF_CONVERGENCE
OPTIONS:
Corresponding to
RECOMMENDATION:
5
HIRSHFELD_READ
Switch to force reading in of isolated atomic densities.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Read in isolated atomic densities from previous Hirshfeld calculation from disk.
FALSE
Generate new isolated atomic densities.
RECOMMENDATION:
Use the default unless system is large. Note, atoms should be in the same order with same basis set used as in the previous Hirshfeld calculation (although coordinates can change). The previous calculation should be run with the -save switch.
HIRSHFELD_SPHAVG
Controls whether atomic densities should be spherically averaged in pro-molecule.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Spherically average atomic densities.
FALSE
Do not spherically average.
RECOMMENDATION:
Use the default.
HIRSHFELD
Controls running of Hirshfeld population analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculate Hirshfeld populations.
FALSE
Do not calculate Hirshfeld populations.
RECOMMENDATION:
None
HIRSHITER_THRESH
Controls the convergence criterion of iterative Hirshfeld population analysis.
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
Corresponding to the convergence criterion of
, in
.
RECOMMENDATION:
Use the default, which is the value recommended in Ref. Bultinck:2007
HIRSHITER
Controls running of iterative Hirshfeld population analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculate iterative Hirshfeld populations.
FALSE
Do not calculate iterative Hirshfeld populations.
RECOMMENDATION:
None
HIRSHMOD
Apply modifiers to the free-atom volumes used in the calculation of the scaled TS-vdW parameters
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
0
Do not apply modifiers to the Hirshfeld volumes.
1
Apply built-in modifier to H.
2
Apply built-in modifier to H and C.
3
Apply built-in modifier to H, C and N.
4
Apply built-in modifier to H, C, N and O
RECOMMENDATION:
Use the default
IDERIV
Controls the order of derivatives that are evaluated analytically. The user is not normally required to specify a value, unless numerical derivatives are desired. The derivatives will be evaluated numerically if IDERIV is set lower than JOBTYPE requires.
TYPE:
INTEGER
DEFAULT:
Set to the order of derivative that JOBTYPE requires
OPTIONS:
2
Analytic second derivatives of the energy (Hessian)
1
Analytic first derivatives of the energy.
0
Analytic energies only.
RECOMMENDATION:
Usually set to the maximum possible for efficiency. Note that IDERIV will be set lower if analytic derivatives of the requested order are not available.
IGDEFIELD
Triggers the calculation of the electrostatic potential and/or the electric field at the positions of the MM charges.
TYPE:
INTEGER
DEFAULT:
UNDEFINED
OPTIONS:
O
Computes ESP.
1
Computes ESP and EFIELD.
2
Computes EFIELD.
RECOMMENDATION:
Must use this $rem when IGDESP is specified.
IGDESP
Controls evaluation of the electrostatic potential on a grid of points. If enabled, the output is in an ASCII file, plot.esp, in the format
esp for each point.
TYPE:
INTEGER
DEFAULT:
none no electrostatic potential evaluation
OPTIONS:

same as the option ’-1’, plus evaluate the ESP of $external_charges$
read grid input via the $plots section of the input deck
Generate the ESP values at all nuclear positions
+
read
RECOMMENDATION:
None
IGNORE_LOW_FREQ
Low frequencies that should be treated as rotation can be ignored during
anharmonic correction calculation.
TYPE:
INTEGER
DEFAULT:
300
Corresponding to 300 cm
.
OPTIONS:
Any mode with harmonic frequency less than
RECOMMENDATION:
Use the default.
INCDFT_DENDIFF_THRESH
Sets the threshold for screening density matrix values in the IncDFT procedure.
TYPE:
INTEGER
DEFAULT:
SCF_CONVERGENCE + 3
OPTIONS:
Corresponding to a threshold of
RECOMMENDATION:
If the default value causes convergence problems, set this value higher to tighten the threshold.
INCDFT_DENDIFF_VARTHRESH
Sets the lower bound for the variable threshold for screening density matrix values in the IncDFT procedure. The threshold will begin at this value and then vary depending on the error in the current SCF iteration until the value specified by INCDFT_DENDIFF_THRESH is reached. This means this value must be set lower than INCDFT_DENDIFF_THRESH.
TYPE:
INTEGER
DEFAULT:
0
Variable threshold is not used.
OPTIONS:
Corresponding to a threshold of
RECOMMENDATION:
If the default value causes convergence problems, set this value higher to tighten accuracy. If this fails, set to 0 and use a static threshold.
INCDFT_GRIDDIFF_THRESH
Sets the threshold for screening functional values in the IncDFT procedure
TYPE:
INTEGER
DEFAULT:
SCF_CONVERGENCE + 3
OPTIONS:
Corresponding to a threshold of
RECOMMENDATION:
If the default value causes convergence problems, set this value higher to tighten the threshold.
INCDFT_GRIDDIFF_VARTHRESH
Sets the lower bound for the variable threshold for screening the functional values in the IncDFT procedure. The threshold will begin at this value and then vary depending on the error in the current SCF iteration until the value specified by INCDFT_GRIDDIFF_THRESH is reached. This means that this value must be set lower than INCDFT_GRIDDIFF_THRESH.
TYPE:
INTEGER
DEFAULT:
0
Variable threshold is not used.
OPTIONS:
Corresponding to a threshold of
RECOMMENDATION:
If the default value causes convergence problems, set this value higher to tighten accuracy. If this fails, set to 0 and use a static threshold.
INCDFT
Toggles the use of the IncDFT procedure for DFT energy calculations.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
FALSE
Do not use IncDFT
TRUE
Use IncDFT
RECOMMENDATION:
Turning this option on can lead to faster SCF calculations, particularly towards the end of the SCF. Please note that for some systems use of this option may lead to convergence problems.
INCFOCK
Iteration number after which the incremental Fock matrix algorithm is initiated
TYPE:
INTEGER
DEFAULT:
1
Start INCFOCK after iteration number 1
OPTIONS:
User-defined (0 switches INCFOCK off)
RECOMMENDATION:
May be necessary to allow several iterations before switching on INCFOCK.
INTEGRALS_BUFFER
Controls the size of in-core integral storage buffer.
TYPE:
INTEGER
DEFAULT:
15
15 Megabytes.
OPTIONS:
User defined size.
RECOMMENDATION:
Use the default, or consult your systems administrator for hardware limits.
INTEGRAL_2E_OPR
Determines the two-electron operator.
TYPE:
INTEGER
DEFAULT:
-2
Coulomb Operator.
OPTIONS:
-1
Apply the CASE approximation.
-2
Coulomb Operator.
RECOMMENDATION:
Use the default unless the CASE operator is desired.
INTERNAL_STABILITY_CONV
Convergence criterion for the Davidson solver (for the lowest eigenvalues).
TYPE:
INTEGER
DEFAULT:
4 (3 when FD_MAT_ON_VECS = TRUE)
OPTIONS:
Terminate Davidson iterations when the norm of the residual vector is below 10
RECOMMENDATION:
Use the default.
INTERNAL_STABILITY_DAVIDSON_ITER
Maximum number of Davidson iterations allowed in one stability analysis.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
Perform up to
RECOMMENDATION:
Use the default.
INTERNAL_STABILITY_ITER
Maximum number of new SCF calculations permitted after the first stability analysis is performed.
TYPE:
INTEGER
DEFAULT:
0 (automatically set to 1 if INTERNAL_STABILITY = TRUE)
OPTIONS:
RECOMMENDATION:
Give a larger number if 1 is not enough (still unstable).
INTERNAL_STABILITY_ROOTS
Number of lowest Hessian eigenvalues to solve for.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
Solve for
RECOMMENDATION:
Use the default.
INTERNAL_STABILITY
Perform internal stability analysis in GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform internal stability analysis after convergence.
TRUE
Perform internal stability analysis and generate the corrected MOs.
RECOMMENDATION:
Turn it on when the SCF solution is prone to unstable solutions, especially for open-shell species.
INTRACULE
Controls whether intracule properties are calculated (see also the $intracule section).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
No intracule properties.
TRUE
Evaluate intracule properties.
RECOMMENDATION:
None
IP_STATES
Sets the number of ionized target states roots to find. By default,
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
0
Do not look for any IP states.
OPTIONS:
Find
RECOMMENDATION:
None
ISOTOPES
Specifies if non-default masses are to be used in the frequency calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Use default masses only.
TRUE
Read isotope masses from $isotopes section.
RECOMMENDATION:
None
JOBTYPE
Specifies the calculation.
TYPE:
STRING
DEFAULT:
Default is single-point, which should be changed to one of the following options.
OPTIONS:
OPT
Equilibrium structure optimization.
TS
Transition structure optimization.
RPATH
Intrinsic reaction path following.
RECOMMENDATION:
Application-dependent.
KS_GAP_PRINT
Control printing of (generalized Kohn-Sham) HOMO-LUMO gap information.
TYPE:
Boolean
DEFAULT:
false
OPTIONS:
false
(default) do not print gap information
true
print gap information
RECOMMENDATION:
Use in conjunction with KS_GAP_UNIT if true.
KS_GAP_UNIT
Unit for KS_GAP_PRINT and FOA_FUNDGAP (see Section 5.11)
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
(default) hartrees
1
eV
RECOMMENDATION:
none
LB94_BETA
Sets the
TYPE:
INTEGER
DEFAULT:
500
OPTIONS:
Corresponding to
.
RECOMMENDATION:
Use the default.
LINK_ATOM_PROJECTION
Controls whether to perform a link-atom projection
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Performs the projection
FALSE
No projection
RECOMMENDATION:
Necessary in a full QM/MM Hessian evaluation on a system with link atoms
LIN_K
Controls whether linear scaling evaluation of exact exchange (LinK) is used.
TYPE:
LOGICAL
DEFAULT:
Program chooses, switching on LinK whenever CFMM is used.
OPTIONS:
TRUE
Use LinK
FALSE
Do not use LinK
RECOMMENDATION:
Use for HF and hybrid DFT calculations with large numbers of atoms.
LOBA_THRESH
Specifies the thresholds to use for LOBA
TYPE:
INTEGER
DEFAULT:
6015
OPTIONS:

specifies the threshold to use for occupation
Both are given as percentages.
RECOMMENDATION:
Decrease
LOBA
Specifies the methods to use for LOBA
TYPE:
INTEGER
DEFAULT:
00
OPTIONS:


specifies the localization method
0 Perform Boys localization.
1 Perform PM localization.
2 Perform ER localization.
specifies the population analysis method
0 Do not perform LOBA. This is the default.
1 Use Mulliken population analysis.
2 Use Löwdin population analysis.
RECOMMENDATION:
Boys Localization is the fastest. ER will require an auxiliary basis set.
LOBA 12 provides a reasonable speed/accuracy compromise.
LOCALFREQ_GROUP1
Select the number of modes to include in the first subset of modes to localize independently when the keyword LOCALFREQ_GROUPS > 0.
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
User-specified integer.
RECOMMENDATION:
Modes will be included starting with the lowest frequency mode and then in ascending energy order up to the defined value.
LOCALFREQ_GROUPS
Select the number of groups of frequencies to be localized separately within a localized mode calculation. The size of the groups are then controlled using the LOCALFREQ_GROUP1, LOCALFREQ_GROUP2, and LOCALFREQ_GROUP3 keywords.
TYPE:
INTEGER
DEFAULT:
0
Localize all normal modes together.
OPTIONS:
1
Define one subset of modes to localize independently.
2
Define two subsets of modes to localize independently.
3
Define three subsets of modes to localize independently.
RECOMMENDATION:
None
LOCALFREQ_MAX_ITER
Controls the maximum number of mode localization sweeps permitted.
TYPE:
INTEGER
DEFAULT:
200
OPTIONS:
User-specified integer.
RECOMMENDATION:
None
LOCALFREQ_SELECT
Select a subset of normal modes for subsequent anharmonic frequency analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use all normal modes.
OPTIONS:
TRUE
Select a subset of normal modes.
RECOMMENDATION:
None
LOCALFREQ_THRESH
Mode localization is considered converged when the change in the localization criterion is less than
.
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
User-specified integer.
RECOMMENDATION:
None
LOCAL_FREQ
Controls whether a vibrational mode localization calculation is performed.
TYPE:
INTEGER
DEFAULT:
0
Normal mode calculation.
OPTIONS:
1
Localized mode calculation with a Pipek-Mezey like criterion.
2
Localized mode calculation with a Boys like criterion.
RECOMMENDATION:
None
LOCAL_INTERP_ORDER
Controls the order of the B-spline
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
An integer
RECOMMENDATION:
The default value is sufficiently accurate
LOC_CIS_OV_SEPARATE
Decide whether or not to localized the “occupied” and “virtual” components of the localized diabatization function, i.e., whether to localize the electron attachments and detachments separately.
TYPE:
LOGICAL
DEFAULT:
FALSE
Do not separately localize electron attachments and detachments.
OPTIONS:
TRUE
RECOMMENDATION:
If one wants to use Boys localized diabatization for energy transfer (as opposed to electron transfer) , this is a necessary option. ER is more rigorous technique, and does not require this OV feature, but will be somewhat slower.
LOWDIN_POPULATION
Run Löwdin population analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not calculate Löwdin populations.
TRUE
Run Löwdin population analysis.
RECOMMENDATION:
None
LRC_DFT
Controls the application of long-range-corrected DFT
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Do not apply long-range correction.
TRUE (or 1)
Add 100% long-range Hartree-Fock exchange to the requested functional.
RECOMMENDATION:
The $rem variable OMEGA must also be specified, in order to set the range-separation parameter.
MAGNET
Activate the magnetic property module.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Don’t activate the magnetic property module.
TRUE (or 1)
Activate the magnetic property module.
RECOMMENDATION:
None.
MANY_BODY_BSSE
Controls the type of many-body BSSE corrections.
TYPE:
STRING
DEFAULT:
MBCP
OPTIONS:
MBCP
Use many-body counterpoise correction.
VMFC
Use Valiron-Mayer function counterpoise correction.
RECOMMENDATION:
NONE.
MANY_BODY_INT
Perform a MBE calculation.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform a MBE calculation.
FALSE
Do not perform a MBE calculation.
RECOMMENDATION:
NONE
MAX_CIS_CYCLES
Maximum number of CIS iterative cycles allowed.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
User-defined number of cycles.
RECOMMENDATION:
Default is usually sufficient.
MAX_CIS_SUBSPACE
Maximum number of subspace vectors allowed in the CIS iterations
TYPE:
INTEGER
DEFAULT:
As many as required to converge all roots
OPTIONS:
User-defined number of subspace vectors
RECOMMENDATION:
The default is usually appropriate, unless a large number of states are requested for a large molecule. The total memory required to store the subspace vectors is bounded above by
, where
and
represent the number of occupied and virtual orbitals, respectively.
MAX_DIIS_CYCLES
The maximum number of DIIS iterations before switching to (geometric) direct minimization when SCF_ALGORITHM is DIIS_GDM or DIIS_DM. See also THRESH_DIIS_SWITCH.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
1
Only a single Roothaan step before switching to (G)DM
RECOMMENDATION:
None
MAX_RCA_CYCLES
The maximum number of RCA iterations before switching to DIIS when SCF_ALGORITHM is RCA_DIIS.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
N
N RCA iterations before switching to DIIS
RECOMMENDATION:
None
MAX_SCF_CYCLES
Controls the maximum number of SCF iterations permitted.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
RECOMMENDATION:
Increase for slowly converging systems such as those containing transition metals.
MBDVDW
Flag to switch on the MBD-vdW method
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not calculate MBD.
1
Calculate the MBD-vdW contribution to the energy.
2
Calculate the MBD-vdW contribution to the energy and the gradient.
RECOMMENDATION:
NONE
MBE_BSSE_ORDER
Controls the order of many-body BSSE corrections.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
Order of many-body BSSE corrections
RECOMMENDATION:
MBCP and VMFC can be performed up to fourth order.
MBE_EMBEDDING
Controls the type of MBE calculations.
TYPE:
STRING
DEFAULT:
GAS
OPTIONS:
GAS
MBE without charge embedding.
CHARGES
MBE with charge embedding.
RECOMMENDATION:
NONE.
MBE_ORDER
Controls the truncation order
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
Order of MBE
RECOMMENDATION:
MBE can be performed up to fifth order.
MECP_METHODS
Determines which method to be used.
TYPE:
STRING
DEFAULT:
BRANCHING_PLANE
OPTIONS:
BRANCHING_PLANE
Use the branching-plane updating method.
MECP_DIRECT
Use the direct method.
PENALTY_FUNCTION
Use the penalty-constrained method.
RECOMMENDATION:
The direct method is stable for small molecules or molecules with high symmetry. The branching-plane updating method is more efficient for larger molecules but does not work if the two states have different symmetries. If using the branching-plane updating method, GEOM_OPT_COORDS must be set to 0 in the $rem section, as this algorithm is available in Cartesian coordinates only. The penalty-constrained method converges slowly and is suggested only if other methods fail.
MECP_OPT
Determines whether we are doing MECP optimizations.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Do MECP optimization.
FALSE
Do not do MECP optimization.
RECOMMENDATION:
None.
MECP_PROJ_HESS
Determines whether to project out the coupling vector from the Hessian when using branching plane updating method.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
FALSE
RECOMMENDATION:
Use the default.
MECP_STATE1
Sets the first Born-Oppenheimer state for MECP optimization.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
None
OPTIONS:
[
Find the
means the SCF ground state.
RECOMMENDATION:
MECP_STATE2
Sets the second Born-Oppenheimer state for MECP optimization.
TYPE:
INTEGER/INTEGER ARRAY
DEFAULT:
None
OPTIONS:
[
Find the
RECOMMENDATION:
MEM_STATIC
Sets the memory for AO-integral evaluations and their transformations.
TYPE:
INTEGER
DEFAULT:
64
corresponding to 64 Mb.
OPTIONS:
User-defined number of megabytes.
RECOMMENDATION:
For RI-MP2 calculations,
of MEM_STATIC is required. Because a number of matrices with
size also need to be stored, 32–160 Mb of additional MEM_STATIC is needed.
MEM_TOTAL
Sets the total memory available to Q-Chem, in megabytes.
TYPE:
INTEGER
DEFAULT:
2000
2 Gb
OPTIONS:
User-defined number of megabytes.
RECOMMENDATION:
Use the default, or set to the physical memory of your machine. The minimum requirement is
.
METECO
Sets the threshold criteria for discarding shell-pairs.
TYPE:
INTEGER
DEFAULT:
2
Discard shell-pairs below
.
OPTIONS:
1
Discard shell-pairs four orders of magnitude below machine precision.
2
Discard shell-pairs below 10
.
RECOMMENDATION:
Use the default.
METHOD
Specifies the exchange-correlation functional.
TYPE:
STRING
DEFAULT:
No default
OPTIONS:
NAME
Use METHOD = NAME, where NAME is either HF for Hartree-Fock theory or
else one of the DFT methods listed in Section 5.3.4.
RECOMMENDATION:
In general, consult the literature to guide your selection. Our recommendations for DFT are indicated in bold in Section 5.3.4.
MGC_AMODEL
Choice of approximate cluster model.
TYPE:
INTEGER
DEFAULT:
Determines
how the CC equations are approximated:
OPTIONS:
0
Local Active-Space Amplitude iterations (pre-calculate GVB orbitals with your method of choice (RPP is good)).
7
Optimize-Orbitals using the VOD 2-step solver.
(Experimental-only use with MGC_AMPS = 2, 24 ,246)
8
Traditional Coupled Cluster up to CCSDTQPH.
9
MR-CC version of the Pair-Models. (Experimental)
RECOMMENDATION:
None
MGC_AMPS
Choice of Amplitude Truncation
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
2
n
which will be iterated. Choose 1234 for PQ and 123456 for PH
RECOMMENDATION:
None
MGC_LOCALINTER
Pair filter on an intermediate.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Any nonzero value enforces the pair constraint on intermediates,
significantly reducing computational cost. Not recommended for
RECOMMENDATION:
None
MGC_LOCALINTS
Pair filter on an integrals.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
Enforces a pair filter on the 2-electron integrals, significantly
reducing computational cost. Generally useful. for more than 1 pair locality.
RECOMMENDATION:
None
MGC_NLPAIRS
Number of local pairs on an amplitude.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
Must be greater than 1, which corresponds to the PP model. 2 for PQ, and 3 for PH.
RECOMMENDATION:
None
MGEMM_THRESH
Sets MGEMM threshold to determine the separation between “large” and “small” matrix elements. A larger threshold value will result in a value closer to the single-precision result. Note that the desired factor should be multiplied by 10000 to ensure an integer value.
TYPE:
INTEGER
DEFAULT:
10000
(corresponds to 1)
OPTIONS:
User-specified threshold
RECOMMENDATION:
For small molecules and basis sets up to triple-
MI_ACTIVE_FRAGMENT
Sets the active fragment
TYPE:
INTEGER
DEFAULT:
NO DEFAULT
OPTIONS:
Specify the fragment on which the TDDFT calculation is to be performed, for LEA-TDDFT(MI).
RECOMMENDATION:
None
MI_LEA
Controls the LEA-TDDFT(MI) methods
TYPE:
INTEGER
DEFAULT:
NO DEFAULT
OPTIONS:
0
The LEA0 method
1
The LEA-Q method
2
The LEAc method
RECOMMENDATION:
1
MM_CHARGES
Requests the calculation of multipole-derived charges (MDCs).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Calculates the MDCs and also the traceless form of the multipole moments
RECOMMENDATION:
Set to TRUE if MDCs or the traceless form of the multipole moments are desired. The calculation does not take long.
MM_SUBTRACTIVE
Specifies whether a subtractive scheme is used in the
, Eq. eq:ECoul, portion of the calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Only pairs that are not 1-2, 1-3, or 1-4 pairs are used.
TRUE
All pairs are calculated, and then the pairs that are double counted (1-2, 1-3, and 1-4) are subtracted out.
RECOMMENDATION:
When running QM/MM or MM calculations there is not recommendation. When running a QM/MM Ewald calculation the value must be set to TRUE.
MODEL_SYSTEM_CHARGE
Specifies the QM subsystem charge if different from the $molecule section.
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
The charge of the QM subsystem.
RECOMMENDATION:
This option only needs to be used if the QM subsystem (model system) has a charge that is different from the total system charge.
MODEL_SYSTEM_MULT
Specifies the QM subsystem multiplicity if different from the $molecule section.
TYPE:
INTEGER
DEFAULT:
NONE
OPTIONS:
The multiplicity of the QM subsystem.
RECOMMENDATION:
This option only needs to be used if the QM subsystem (model system) has a multiplicity that is different from the total system multiplicity. ONIOM calculations must be closed shell.
MODE_COUPLING
Number of modes coupling in the third and fourth derivatives calculation.
TYPE:
INTEGER
DEFAULT:
2
for two modes coupling.
OPTIONS:
for
RECOMMENDATION:
Use the default.
MOLDEN_FORMAT
Requests a MolDen-formatted input file containing information from a Q-Chem job.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Append MolDen input file at the end of the Q-Chem output file.
RECOMMENDATION:
None.
MOM_METHOD
Determines the target orbitals with which to maximize the overlap on each SCF cycle.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
3
Maximize overlap with the orbitals from the previous SCF cycle.
13
Maximize overlap with the initial guess orbitals.
RECOMMENDATION:
If appropriate guess orbitals can be obtained, then MOM_METHOD = 13 can provide more reliable convergence to the desired solution.
MOM_PRINT
Switches printing on within the MOM procedure.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Printing is turned off
TRUE
Printing is turned on.
RECOMMENDATION:
None
MOM_START
Determines when MOM is switched on to stabilize DIIS iterations.
TYPE:
INTEGER
DEFAULT:
0 (FALSE)
OPTIONS:
0 (FALSE)
MOM is not used
MOM begins on cycle
RECOMMENDATION:
Set to 1 if preservation of initial orbitals is desired. If MOM is to be used to aid convergence, an SCF without MOM should be run to determine when the SCF starts oscillating. MOM should be set to start just before the oscillations.
MOPROP_CONV_1ST
Sets the convergence criteria for CPSCF and 1st order TDSCF.
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
Convergence threshold set to
RECOMMENDATION:
None
MOPROP_CONV_2ND
Sets the convergence criterion for second-order TDSCF.
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
Convergence threshold set to
RECOMMENDATION:
None
MOPROP_DIIS_DIM_SS
Specified the DIIS subspace dimension.
TYPE:
INTEGER
DEFAULT:
20
OPTIONS:
0
No DIIS.
Use a subspace of dimension
RECOMMENDATION:
None
MOPROP_DIIS
Controls the use of Pulay’s DIIS in solving the CPSCF equations.
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
0
Turn off DIIS.
5
Turn on DIIS.
RECOMMENDATION:
None
MOPROP_ISSC_PRINT_REDUCED
Specifies whether the isotope-independent reduced coupling tensor
should be printed in addition to the isotope-dependent
-tensor when calculating indirect nuclear spin-spin couplings.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not print
TRUE
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_DSO
Specifies whether to skip the calculation of the diamagnetic spin-orbit contribution to the indirect nuclear spin-spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate diamagnetic spin-orbit contribution.
TRUE
Skip diamagnetic spin-orbit contribution.
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_FC
Specifies whether to skip the calculation of the Fermi contact contribution to the indirect nuclear spin-spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate Fermi contact contribution.
TRUE
Skip Fermi contact contribution.
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_PSO
Specifies whether to skip the calculation of the paramagnetic spin-orbit contribution to the indirect nuclear spin-spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate paramagnetic spin-orbit contribution.
TRUE
Skip paramagnetic spin-orbit contribution.
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_SD
Specifies whether to skip the calculation of the spin-dipole contribution to the indirect nuclear spin-spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate spin-dipole contribution.
TRUE
Skip spin-dipole contribution.
RECOMMENDATION:
None
MOPROP_MAXITER_1ST
The maximum number of iterations for CPSCF and first-order TDSCF.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
Set maximum number of iterations to
RECOMMENDATION:
Use the default.
MOPROP_MAXITER_2ND
The maximum number of iterations for second-order TDSCF.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
Set maximum number of iterations to
RECOMMENDATION:
Use the default.
MOPROP_PERTNUM
Set the number of perturbed densities that will to be treated together.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
All at once.
Treat the perturbed densities batch-wise.
RECOMMENDATION:
Use the default. For large systems, limiting this number may be required to avoid memory exhaustion.
MOPROP_RESTART
Specifies the option for restarting MOProp calculations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Not a restart calculation.
1
Restart from a previous calculation using the same scratch directory.
RECOMMENDATION:
Need to also include "SCF_GUESS READ" and "SKIP_SCFMAN TRUE" to ensure the same set of MOs.
MOPROP
Specifies the job number for MOProp module.
TYPE:
INTEGER
DEFAULT:
0
Do not run the MOProp module.
OPTIONS:
1
NMR chemical shielding tensors.
2
Static polarizability.
3
Indirect nuclear spin–spin coupling tensors.
100
Dynamic polarizability.
101
First hyperpolarizability.
102
First hyperpolarizability, reading First order results from disk.
103
First hyperpolarizability using Wigner’s
rule.
104
First hyperpolarizability using Wigner’s
first order results from disk.
RECOMMENDATION:
None
MRXC_CLASS_THRESH_MULT
Controls the of smoothness precision
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
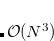
An integer
RECOMMENDATION:
A prefactor in the threshold for MRXC error control:
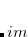
MRXC_CLASS_THRESH_ORDER
Controls the of smoothness precision
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
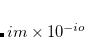
An integer
RECOMMENDATION:
The exponent in the threshold of the MRXC error control:
MRXC
Controls the use of MRXC.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not use MRXC
1
Use MRXC in the evaluation of the XC part
RECOMMENDATION:
MRXC is very efficient for medium and large molecules, especially when medium and large basis sets are used.
MULTIPOLE_ORDER
Determines highest order of multipole moments to print if wave function analysis requested.
TYPE:
INTEGER
DEFAULT:
4
OPTIONS:
Calculate moments to
RECOMMENDATION:
Use the default unless higher multipoles are required.
NBO
Controls the use of the NBO package.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not invoke the NBO package.
1
Do invoke the NBO package, for the ground state.
2
Invoke the NBO package for the ground state, and also each
CIS, RPA, or TDDFT excited state.
RECOMMENDATION:
None
NL_CORRELATION
Specifies a non-local correlation functional that includes non-empirical dispersion.
TYPE:
STRING
DEFAULT:
None
No non-local correlation.
OPTIONS:
None
No non-local correlation
vdW-DF-04
the non-local part of vdW-DF-04
vdW-DF-10
the non-local part of vdW-DF-10 (also known as vdW-DF2)
VV09
the non-local part of VV09
VV10
the non-local part of VV10
RECOMMENDATION:
Do not forget to add the LSDA correlation (PW92 is recommended) when using vdW-DF-04, vdW-DF-10, or VV09. VV10 should be used with PBE correlation. Choose exchange functionals carefully: HF, rPW86, revPBE, and some of the LRC exchange functionals are among the recommended choices.
NL_GRID
Specifies the grid to use for non-local correlation.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
Same as for XC_GRID
RECOMMENDATION:
Use the default unless computational cost becomes prohibitive, in which case SG-0 may be used. XC_GRID should generally be finer than NL_GRID.
NL_VV_B
Sets the parameter
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to
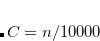
RECOMMENDATION:
The optimal value depends strongly on the exchange functional used.
is recommended for rPW86. For further details see Ref. Vydrov:2010b.
NL_VV_C
Sets the parameter
in VV09 and VV10. This parameter is fitted to asymptotic van der Waals
TYPE:
INTEGER
DEFAULT:
89
for VV09
No default
for VV10
OPTIONS:
Corresponding to
RECOMMENDATION:
is recommended when a semi-local exchange functional is used.
is recommended when a long-range corrected (LRC) hybrid functional is used. For further details see Ref. Vydrov:2010b.
NOCI_PRINT
Specify the debug print level of NOCI.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
Positive integer
RECOMMENDATION:
Increase this for additional debug information.
NOSE_HOOVER_LENGTH
Sets the chain length for the Nosé-Hoover thermostat
TYPE:
INTEGER
DEFAULT:
none
OPTIONS:
Chain length of
RECOMMENDATION:
Typically 3-6
NOSE_HOOVER_TIMESCALE
Sets the timescale (strength) of the Nosé-Hoover thermostat
TYPE:
INTEGER
DEFAULT:
none
OPTIONS:
Thermostat timescale, as
RECOMMENDATION:
Smaller values (roughly 100) equate to tighter thermostats but may inhibit rapid sampling. Larger values (
NTO_PAIRS
Controls the writing of hole/particle NTO pairs for excited state.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Write
RECOMMENDATION:
If activated (
NVO_LIN_CONVERGENCE
Target error factor in the preconditioned conjugate gradient solver of the single-excitation amplitude equations.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
User–defined number.
RECOMMENDATION:
Solution of the single-excitation amplitude equations is considered converged if the maximum residual is less than
NVO_LIN_MAX_ITE
Maximum number of iterations in the preconditioned conjugate gradient solver of the single-excitation amplitude equations.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
User–defined number of iterations.
RECOMMENDATION:
None.
NVO_METHOD
Sets method to be used to converge solution of the single-excitation amplitude equations.
TYPE:
INTEGER
DEFAULT:
9
OPTIONS:
User–defined number.
RECOMMENDATION:
This is an experimental option. Use the default.
NVO_TRUNCATE_DIST
Specifies which atomic blocks of the Fock matrix are used to construct the preconditioner.
TYPE:
INTEGER
DEFAULT:
-1
OPTIONS:
If distance between a pair of atoms is more than
do not include the atomic block.
-2
Do not use distance threshold, use NVO_TRUNCATE_PRECOND instead.
-1
Include all blocks.
0
Include diagonal blocks only.
RECOMMENDATION:
This option does not affect the final result. However, it affects the rate of the PCG algorithm convergence. For small systems, use the default.
NVO_TRUNCATE_PRECOND
Specifies which atomic blocks of the Fock matrix are used to construct the preconditioner. This variable is used only if NVO_TRUNCATE_DIST is set to
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
If the maximum element in an atomic block is less than
the block.
RECOMMENDATION:
Use the default. Increasing
NVO_UVV_MAXPWR
Controls convergence of the Taylor series when calculating the
block from the single-excitation amplitudes. If the series is not converged at the
TYPE:
INTEGER
DEFAULT:
10
OPTIONS:
User–defined number.
RECOMMENDATION:
None.
NVO_UVV_PRECISION
Controls convergence of the Taylor series when calculating the
TYPE:
INTEGER
DEFAULT:
11
OPTIONS:
User–defined number.
RECOMMENDATION:
NVO_UVV_PRECISION must be the same as or larger than THRESH.
N_FROZEN_CORE
Sets the number of frozen core orbitals in a post-Hartree–Fock calculation.
TYPE:
INTEGER
DEFAULT:
FC
OPTIONS:
FC
Frozen Core approximation (all core orbitals frozen).
Freeze
RECOMMENDATION:
Correlated calculations calculations are more efficient with frozen core orbitals. Use default if possible.
N_FROZEN_VIRTUAL
Sets the number of frozen virtual orbitals in a post-Hartree–Fock calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Freeze
RECOMMENDATION:
None
N_I_SERIES
Sets summation limit for series expansion evaluation of
.
TYPE:
INTEGER
DEFAULT:
40
OPTIONS:
RECOMMENDATION:
Lower values speed up the calculation, but may affect accuracy.
N_J_SERIES
Sets summation limit for series expansion evaluation of
.
TYPE:
INTEGER
DEFAULT:
40
OPTIONS:
RECOMMENDATION:
Lower values speed up the calculation, but may affect accuracy.
N_SOL
Specifies number of atoms included in the Hessian.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
User defined
RECOMMENDATION:
None
N_WIG_SERIES
Sets summation limit for Wigner integrals.
TYPE:
INTEGER
DEFAULT:
10
OPTIONS:

RECOMMENDATION:
Increase
OCCUPATIONS
Activates pFON calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Integer occupation numbers
1
Not yet implemented
2
Pseudo-fractional occupation numbers (pFON)
RECOMMENDATION:
Use pFON to improve convergence for small-gap systems.
OCC_RI_K
Controls the use of the occ-RI-K approximation for constructing the exchange matrix
TYPE:
LOGICAL
DEFAULT:
False
Do not use occ-RI-K.
OPTIONS:
True
Use occ-RI-K.
RECOMMENDATION:
Larger the system, better the performance
OMEGA2
Sets the Coulomb attenuation parameter for the long-range component.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to
, in units of bohr
RECOMMENDATION:
None
OMEGA_GDD_SCALING
Sets the empirical constant
tuning procedure.
TYPE:
INTEGER
DEFAULT:
885
OPTIONS:
Corresponding to
.
RECOMMENDATION:
The quantity
OMEGA_GDD
Controls the application of
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Do not apply
TRUE (or 1)
Use
RECOMMENDATION:
The $rem variable OMEGA must also be specified, in order to set the initial range-separation parameter.
OMEGA
Controls the degree of attenuation of the Coulomb operator.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to
, in units of bohr
RECOMMENDATION:
None
OMEGA
Sets the Coulomb attenuation parameter for the short-range component.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
Corresponding to
RECOMMENDATION:
None
OS_ROSCF
Run an open-shell singlet ROSCF calculation with GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
OS_ROSCF calculation is performed.
FALSE
Do not run OS_ROSCF (it will run a close-shell RSCF calculation instead).
RECOMMENDATION:
Set to TRUE if desired.
PAO_ALGORITHM
Algorithm used to optimize polarized atomic orbitals (see PAO_METHOD)
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use efficient (and riskier) strategy to converge PAOs.
1
Use conservative (and slower) strategy to converge PAOs.
RECOMMENDATION:
None
PAO_METHOD
Controls evaluation of polarized atomic orbitals (PAOs).
TYPE:
STRING
DEFAULT:
EPAO
For local MP2 calculations Otherwise no default.
OPTIONS:
PAO
Perform PAO-SCF instead of conventional SCF.
EPAO
Obtain EPAOs after a conventional SCF.
RECOMMENDATION:
None
PARI_K
Controls the use of the PARI-K approximation in the construction of the exchange matrix
TYPE:
LOGICAL
DEFAULT:
FALSE
Do not use PARI-K.
OPTIONS:
TRUE
Use PARI-K.
RECOMMENDATION:
Use for basis sets aug-cc-pVTZ and larger.
PBHT_ANALYSIS
Controls whether overlap analysis of electronic excitations is performed.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform overlap analysis.
TRUE
Perform overlap analysis.
RECOMMENDATION:
None
PBHT_FINE
Increases accuracy of overlap analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
TRUE
Increase accuracy of overlap analysis.
RECOMMENDATION:
None
PEQ_SWITCH
Inclusion of solvent effects begins when the SCF error falls below 10
.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
Corresponding to 10
RECOMMENDATION:
Use the default unless solvent effects need to be incorporated earlier in the SCF procedure.
PHESS
Controls whether partial Hessian calculations are performed.
TYPE:
INTEGER
DEFAULT:
0
Full Hessian calculation
OPTIONS:
1
Partial Hessian calculation.
2
Vibrational subsystem analysis (massless).
3
Vibrational subsystem analysis (weighted).
RECOMMENDATION:
None
PH_FAST
Lowers integral cutoff in partial Hessian calculation is performed.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use default cutoffs
OPTIONS:
TRUE
Lower integral cutoffs
RECOMMENDATION:
None
PIMC_ACCEPT_RATE
Acceptance rate for MC/PIMC simulations when Cartesian or normal-mode displacements are used.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:

User-specified rate, given as a whole-number percentage.
RECOMMENDATION:
Choose acceptance rate to maximize sampling efficiency, which is typically signified by the mean-square displacement (printed in the job output). Note that the maximum displacement is adjusted during the warm-up run to achieve roughly this acceptance rate.
PIMC_MCMAX
Number of Monte Carlo steps to sample.
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
User-specified number of steps to sample.
RECOMMENDATION:
This variable dictates the statistical convergence of MC/PIMC simulations. For converged simulations at least
steps is recommended.
PIMC_MOVETYPE
Selects the type of displacements used in MC/PIMC simulations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Cartesian displacements of all beads, with occasional (1%) center-of-mass moves.
1
Normal-mode displacements of all modes, with occasional (1%) center-of-mass moves.
2
Levy flights without center-of-mass moves.
RECOMMENDATION:
Except for classical sampling (MC) or small bead-number quantum sampling (PIMC), Levy flights should be used. For Cartesian and normal-mode moves, the maximum displacement is adjusted during the warm-up run to the desired acceptance rate (controlled by PIMC_ACCEPT_RATE). For Levy flights, the acceptance is solely controlled by PIMC_SNIP_LENGTH.
PIMC_NBEADSPERATOM
Number of path integral time slices (“beads”) used on each atom of a PIMC simulation.
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
1
Perform classical Boltzmann sampling.
1
Perform quantum-mechanical path integral sampling.
RECOMMENDATION:
This variable controls the inherent convergence of the path integral simulation. The one-bead limit represents classical sampling and the infinite-bead limit represents exact quantum-mechanical sampling. Using 32 beads is reasonably converged for room-temperature simulations of molecular systems.
PIMC_SNIP_LENGTH
Number of “beads” to use in the Levy flight movement of the ring polymer.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:

User-specified length of snippet.
RECOMMENDATION:
Choose the snip length to maximize sampling efficiency. The efficiency can be estimated by the mean-square displacement between configurations, printed at the end of the output file. This efficiency will typically, however, be a trade-off between the mean-square displacement (length of statistical correlations) and the number of beads moved. Only the moved beads require recomputing the potential, i.e., a call to Q-Chem for the electronic energy. (Note that the endpoints of the snippet remain fixed during a single move, so
beads are actually moved for a snip length of
PIMC_TEMP
Temperature, in Kelvin (K), of path integral simulations.
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
User-specified number of Kelvin for PIMC or classical MC simulations.
RECOMMENDATION:
None.
PIMC_WARMUP_MCMAX
Number of Monte Carlo steps to sample during an equilibration period of MC/PIMC simulations.
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
User-specified number of steps to sample.
RECOMMENDATION:
Use this variable to equilibrate the molecule/ring polymer before collecting production statistics. Usually a short run of roughly 10% of PIMC_MCMAX is sufficient.
PLOT_SPIN_DENSITY
Requests the generation of spin densities,
and
.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not generate spin density cube files.
TRUE
Generate spin density cube files.
RECOMMENDATION:
Set to TRUE if spin densities are desired in addition to total densities. Requires that MAKE_CUBE_FILES be set to TRUE as well, and that one or more total densities is requested in the $plots input section. The corresponding spin densities will then be generated also.
POL_GEOM
Compute forces on the polarized (converged SCFMI) PES.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not compute forces on the polarized PES.
TRUE
Compute forces on the polarized PES.
RECOMMENDATION:
Set it to TRUE when optimized geometry or vibrational frequencies on the polarized PES are desired.
POP_MULLIKEN
Controls running of Mulliken population analysis.
TYPE:
LOGICAL/INTEGER
DEFAULT:
TRUE (or 1)
OPTIONS:
FALSE (or 0)
Do not calculate Mulliken populations.
TRUE (or 1)
Calculate Mulliken populations.
2
Also calculate shell populations for each occupied orbital.
Calculate Mulliken charges for both the ground state and any CIS,
RPA, or TDDFT excited states.
RECOMMENDATION:
Leave as TRUE, unless excited-state charges are desired. Mulliken analysis is a trivial additional calculation, for ground or excited states.
PRINT_CORE_CHARACTER
Determines the print level for the CORE_CHARACTER option.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No additional output is printed.
1
Prints core characters of occupied MOs.
2
Print level 1, plus prints the core character of AOs.
RECOMMENDATION:
Use the default, unless you are uncertain about what the core character is.
PRINT_DIST_MATRIX
Controls the printing of the inter-atomic distance matrix
TYPE:
INTEGER
DEFAULT:
15
OPTIONS:
0
Turns off the printing of the distance matrix
Prints the distance matrix if the number of atoms in the molecule
is less than or equal to
RECOMMENDATION:
Use default unless distances are required for large systems
PRINT_GENERAL_BASIS
Controls print out of built in basis sets in input format
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Print out standard basis set information
FALSE
Do not print out standard basis set information
RECOMMENDATION:
Useful for modification of standard basis sets.
PRINT_ORBITALS
Prints orbital coefficients with atom labels in analysis part of output.
TYPE:
INTEGER/LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not print any orbitals.
TRUE
Prints occupied orbitals plus 5 virtual orbitals.
NVIRT
Number of virtual orbitals to print.
RECOMMENDATION:
Use true unless more virtual orbitals are desired.
PRINT_RADII_GYRE
Controls printing of MO centroids and radii of gyration.
TYPE:
LOGICAL/INTEGER
DEFAULT:
FALSE
OPTIONS:
TRUE (or 1)
Print the centroid and radius of gyration for each occupied MO and each density.
2
Print centroids and radii of gyration for the virtual MOs as well.
FALSE (or 0)
Do not calculate these quantities.
RECOMMENDATION:
None
PROJ_TRANSROT
Removes translational and rotational drift during AIMD trajectories.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not apply translation/rotation corrections.
TRUE
Apply translation/rotation corrections.
RECOMMENDATION:
When computing spectra (see AIMD_NUCL_DACF_POINTS, for example), this option can be used to remove artificial, contaminating peaks stemming from translational and/or rotational motion. Recommend setting to TRUE for all dynamics-based spectral simulations.
PSEUDO_CANONICAL
When SCF_ALGORITHM = DM, this controls the way the initial step, and steps after subspace resets are taken.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Use Roothaan steps when (re)initializing
TRUE
Use a steepest descent step when (re)initializing
RECOMMENDATION:
The default is usually more efficient, but choosing TRUE sometimes avoids problems with orbital reordering.
PURECART
INTEGER
TYPE:
Controls the use of pure (spherical harmonic) or Cartesian angular forms
DEFAULT:
2111
Cartesian
-functions and pure
functions
OPTIONS:

Use 1 for pure and 2 for Cartesian.
RECOMMENDATION:
This is pre-defined for all standard basis sets
QMMM_CHARGES
Controls the printing of QM charges to file.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Writes a charges.dat file with the Mulliken charges from the QM region.
FALSE
No file written.
RECOMMENDATION:
Use the default unless running calculations with Charmm where charges on the QM region need to be saved.
QMMM_FULL_HESSIAN
Trigger the evaluation of the full QM/MM Hessian.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Evaluates full Hessian.
FALSE
Hessian for QM-QM block only.
RECOMMENDATION:
None
QMMM_PRINT
Controls the amount of output printed from a QM/MM job.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Limit molecule, point charge, and analysis printing.
FALSE
Normal printing.
RECOMMENDATION:
Use the default unless running calculations with Charmm.
QM_MM_INTERFACE
Enables internal QM/MM calculations.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
MM
Molecular mechanics calculation (i.e., no QM region)
ONIOM
QM/MM calculation using two-layer mechanical embedding
JANUS
QM/MM calculation using electronic embedding
RECOMMENDATION:
The ONIOM model and Janus models are described above. Choosing MM leads to no electronic structure calculation. However, when using MM, one still needs to define the $rem variables BASIS and EXCHANGE in order for Q-Chem to proceed smoothly.
QM_MM
Turns on the Q-Chem/Charmm interface.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Do QM/MM calculation through the Q-Chem/Charmm interface.
FALSE
Turn this feature off.
RECOMMENDATION:
Use the default unless running calculations with Charmm.
RAS_ACT_DIFF
Sets the number of alpha vs. beta electrons
TYPE:
Integer
DEFAULT:
None
OPTIONS:
n
user defined integer
RECOMMENDATION:
Set to 1 for an odd number of electrons or a cation, -1 for an anion. Only works with RASCI2.
RAS_ACT_OCC
Sets the number of occupied orbitals to enter the RAS active space.
TYPE:
Integer
DEFAULT:
None
OPTIONS:
n
user defined integer
RECOMMENDATION:
None. Only works with RASCI2
RAS_ACT_ORB
Sets the user-selected active orbitals (RAS2 orbitals).
TYPE:
INTEGER ARRAY
DEFAULT:
From RAS_OCC+1 to RAS_OCC+RAS_ACT
OPTIONS:
![$[i,j,k...]$](images/img-1140.png)
The number of orbitals must be equal to the RAS_ACT variable
RECOMMENDATION:
None. Only works with RASCI.
RAS_ACT_VIR
Sets the number of virtual orbitals to enter the RAS active space.
TYPE:
Integer
DEFAULT:
None
OPTIONS:
n
user defined integer
RECOMMENDATION:
None. Only works with RASCI2.
RAS_ACT
Sets the number of orbitals in RAS2 (active orbitals).
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
User-defined integer,
RECOMMENDATION:
None. Only works with RASCI.
RAS_AMPL_PRINT
Defines the absolute threshold (
) for the CI amplitudes to be printed.
TYPE:
INTEGER
DEFAULT:
10
0.1 minimum absolute amplitude
OPTIONS:
User-defined integer,

RECOMMENDATION:
None. Only works with RASCI.
RAS_DO_HOLE
Controls the presence of hole excitations in the RAS-CI wave function.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Include hole configurations (RAS1 to RAS2 excitations)
FALSE
Do not include hole configurations
RECOMMENDATION:
None. Only works with RASCI.
RAS_DO_PART
Controls the presence of particle excitations in the RAS-CI wave function.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Include particle configurations (RAS2 to RAS3 excitations)
FALSE
Do not include particle configurations
RECOMMENDATION:
None. Only works with RASCI.
RAS_ELEC
Sets the number of electrons in RAS2 (active electrons).
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
User-defined integer,
RECOMMENDATION:
None. Only works with RASCI.
RAS_FRAG_SETS
Sets the number of atoms in each fragment.
TYPE:
INTEGER ARRAY
DEFAULT:
[NOcc,NAct,NVir]
Number of orbitals within RAS1, RAS2 and RAS3 spaces
OPTIONS:
Defines sets of canonical MOs to be localized into
RECOMMENDATION:
Setting within RAS1, RAS2 and RAS3 spaces alleviates the computational cost of the localization procedure. It might also result in improved fragment orbitals. Only works with RASCI.
RAS_GUESS_CS
Controls the number of closed shell guess configurations in RAS-CI.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Imposes to start with
RECOMMENDATION:
Only relevant for the computation of singlet states. Only works with RASCI.
RAS_NATORB_STATE
Allows to save the natural orbitals of a RAS-CI computed state.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Saves the natural orbitals for the
RECOMMENDATION:
None. Only works with RASCI.
RAS_NATORB
Controls the computation of the natural orbital occupancies.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Compute natural orbital occupancies for all states
FALSE
Do not compute natural orbital occupancies
RECOMMENDATION:
None. Only works with RASCI.
RAS_NFRAG_ATOMS
Sets the number of atoms in each fragment.
TYPE:
INTEGER ARRAY
DEFAULT:
None
OPTIONS:
The sum of the numbers must be equal to the total number of atoms in the systems
RECOMMENDATION:
None. Only works with RASCI.
RAS_NFRAG
If
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Number of fragments to be considered
RECOMMENDATION:
Only for RASCI. The printed information level is controlled by RAS_PRINT.
RAS_N_ROOTS
Sets the number of RAS-CI roots to be computed.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
RECOMMENDATION:
None. Only works with RASCI2
RAS_OCC
Sets the number of orbitals in RAS1
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined integer,
RECOMMENDATION:
These are the initial doubly occupied orbitals (RAS1) before including
type of excitations. The RAS1 space starts from the lowest orbital up to RAS_OCC, i.e. no frozen orbitals option available yet. Only works with RASCI.
RAS_PT2_PARTITION
Specifies the partitioning scheme in RASCI(2)
TYPE:
INTEGER
DEFAULT:
1
Davidson-Kapuy (DK) partitioning
OPTIONS:
2
Epstein-Nesbet (EN) partitioning
0
Do both DK and EN partitionings
RECOMMENDATION:
Only for RASCI if RAS_PT2 is set to true.
RAS_PT2_VSHIFT
Defines the energy level shift (
) in RASCI(2)
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined integer
RECOMMENDATION:
Only for RASCI if RAS_PT2 is set to true.
RAS_PT2
Perform second-order perturbative correction to RAS-CI energy
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Compute RASCI(2) energy corrections
FALSE
Do not compute RASCI(2) energy corrections
RECOMMENDATION:
None. Only works with RASCI.
RAS_ROOTS
Sets the number of RAS-CI roots to be computed.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
RECOMMENDATION:
None. Only works with RASCI.
RAS_SPIN_MULT
Specifies the spin multiplicity of the roots to be computed
TYPE:
INTEGER
DEFAULT:
1
Singlet states
OPTIONS:
0
Compute any spin multiplicity
User-defined integer,
RECOMMENDATION:
Only for RASCI, which at present only allows for the computation of systems with an even number of electrons. Thus, RAS_SPIN_MULT only can take odd values.
RCA_PRINT
Controls the output from RCA SCF optimizations.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No print out
1
RCA summary information
2
Level 1 plus RCA coefficients
3
Level 2 plus RCA iteration details
RECOMMENDATION:
None
RC_R0
Determines the parameter in the Gaussian weight function used to smooth the density at the nuclei.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Corresponds the traditional delta function spin and charge densities
corresponding to
a.u.
RECOMMENDATION:
We recommend value of 250 for a typical spit valence basis. For basis sets with increased flexibility in the nuclear vicinity the smaller values of
also yield adequate spin density.
RESPONSE
Activate the general response property module.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE (or 0)
Don’t activate the general response property module.
TRUE (or 1)
Activate the general response property module.
RECOMMENDATION:
None.
RISAPT
Requests an RI-SAPT calculation
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Compute four-index integrals using the RI approximation.
FALSE
Do not use RI.
RECOMMENDATION:
Set to TRUE if an appropriate auxiliary basis set is available, as RI-SAPT is much faster and affords negligible errors (as compared to ordinary SAPT) if the auxiliary basis set is matched to the primary basis set. (The former must be specified using AUX_BASIS.)
RI_J
Toggles the use of the RI algorithm to compute J.
TYPE:
LOGICAL
DEFAULT:
FALSE
RI will not be used to compute J.
OPTIONS:
TRUE
Turn on RI for J.
RECOMMENDATION:
For large (especially 1D and 2D) molecules the approximation may yield significant improvements in Fock evaluation time when used with ARI.
RI_K_GRAD
Turn on the nuclear gradient calculations
TYPE:
LOGICAL
DEFAULT:
FALSE
Do not invoke occ-RI-K based gradient
OPTIONS:
TRUE
Use occ-RI-K based gradient
RECOMMENDATION:
Use "RI_J false"
RI_K
Toggles the use of the RI algorithm to compute K.
TYPE:
LOGICAL
DEFAULT:
FALSE
RI will not be used to compute K.
OPTIONS:
TRUE
Turn on RI for K.
RECOMMENDATION:
For large (especially 1D and 2D) molecules the approximation may yield significant improvements in Fock evaluation time when used with ARI.
ROKS_LEVEL_SHIFT
Introduce a level shift of N/100 hartree to aid convergence.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No shift
N
level shift of N/100 hartree.
RECOMMENDATION:
Use in cases of problematic convergence.
ROKS
Controls whether ROKS calculation will be performed.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
ROKS is not performed.
TRUE
ROKS will be performed.
RECOMMENDATION:
Set to TRUE if ROKS calculation is desired. You should also set UNRESTRICTED = FALSE
RPATH_COORDS
Determines which coordinate system to use in the IRC search.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use mass-weighted coordinates.
1
Use Cartesian coordinates.
2
Use Z-matrix coordinates.
RECOMMENDATION:
Use the default.
RPATH_DIRECTION
Determines the direction of the eigenmode to follow. This will not usually be known prior to the Hessian diagonalization.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Descend in the positive direction of the eigen mode.
-1
Descend in the negative direction of the eigen mode.
RECOMMENDATION:
It is usually not possible to determine in which direction to go a priori, and therefore both directions will need to be considered.
RPATH_MAX_CYCLES
Specifies the maximum number of points to find on the reaction path.
TYPE:
INTEGER
DEFAULT:
20
OPTIONS:
User-defined number of cycles.
RECOMMENDATION:
Use more points if the minimum is desired, but not reached using the default.
RPATH_MAX_STEPSIZE
Specifies the maximum step size to be taken (in 0.001 a.u.).
TYPE:
INTEGER
DEFAULT:
150
corresponding to a step size of 0.15 a.u..
OPTIONS:
Step size =
RECOMMENDATION:
None.
RPATH_PRINT
Specifies the print output level.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
RECOMMENDATION:
Use the default, as little additional information is printed at higher levels. Most of the output arises from the multiple single point calculations that are performed along the reaction pathway.
RPATH_TOL_DISPLACEMENT
Specifies the convergence threshold for the step. If a step size is chosen by the algorithm that is smaller than this, the path is deemed to have reached the minimum.
TYPE:
INTEGER
DEFAULT:
5000
Corresponding to 0.005 a.u.
OPTIONS:
User-defined. Tolerance =
RECOMMENDATION:
Use the default. Note that this option only controls the threshold for ending the RPATH job and does nothing to the intermediate steps of the calculation. A smaller value will provide reaction paths that end closer to the true minimum. Use of smaller values without adjusting RPATH_MAX_STEPSIZE, however, can lead to oscillations about the minimum.
RPA
Do an RPA calculation in addition to a CIS or TDDFT/TDA calculation.
TYPE:
LOGICAL/INTEGER
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not do an RPA calculation.
TRUE
Do an RPA calculation.
2
Do an RPA calculation without running CIS or TDDFT/TDA first.
RECOMMENDATION:
None
MAKE_CUBE_FILES
Requests generation of cube files for MOs, NTOs, or NBOs.
TYPE:
LOGICAL/STRING
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not generate cube files.
TRUE
Generate cube files for MOs and densities.
NTOS
Generate cube files for NTOs.
NBOS
Generate cube files for NBOs.
RECOMMENDATION:
None
SAPT_AO
Request an atomic-orbital version of SAPT.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Use the AO version of SAPT.
FALSE
Use the MO version of SAPT.
RECOMMENDATION:
Use the AO version, which exhibits
scaling without significant memory bottlenecks.
SAPT_BASIS
Controls the MO basis used for SAPT corrections.
TYPE:
STRING
DEFAULT:
MONOMER
OPTIONS:
MONOMER
Monomer-centered basis set (MCBS).
DIMER
Dimer-centered basis set (DCBS).
PROJECTED
Projected (pseudocanonicalized) basis set.
RECOMMENDATION:
The DCBS is more costly than the MCBS and can only be used with XPOL_MPOL_ORDER = GAS (i.e., it is not available for use with XPol). The PROJECTED choice is an efficient compromise that is available for use with XPol.
SAPT_CDFT_EDA
Request a SAPT/cDFT energy decomposition analysis
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Run a SAPT/cDFT calculation.
FALSE
Do not run SAPT/cDFT.
RECOMMENDATION:
None
SAPT_CPHF
Requests that the second-order corrections
and
be replaced by their infinite-order “response” analogues,
and
.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Evaluate the response corrections and use
FALSE
Omit these corrections and use
RECOMMENDATION:
Computing the response corrections requires solving CPHF equations for pair of monomers, which is somewhat expensive but may improve the accuracy when the monomers are polar.
SAPT_DISP_CORR
Request an empirical dispersion potential instead of calculating
and
directly.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Use a dispersion force field.
FALSE
Calculate
RECOMMENDATION:
Dispersion potentials combined with AO-SAPT reduces the scaling from
to
SAPT_DISP_VERSION
Controls which dispersion potential is used for SAPT.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
1
Use the “first generation” (+aiD1) dispersion potential.[Lao and Herbert(2012b)]
2
Use the “second generation” (+aiD2) dispersion potential.[Lao and Herbert(2013)]
3
Use the “third generation” (+aiD3) dispersion potentials.[Lao and Herbert(2015)]
RECOMMENDATION:
Use +aiD3. The second- and third-generation versions were parameterized using ab initio dispersion data and afford accurate energy components, in addition to accurate total interaction energies. The third-generation version was parameterized using an expanded data set designed to reduce some large errors observed for
-stacked complexes using +aiD2.
SAPT_DSCF
Request the
correction
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Evaluate this correction.
FALSE
Omit this correction.
RECOMMENDATION:
Evaluating the
SAPT_EXCHANGE
Selects the type of first-order exchange that is used in a SAPT calculation.
TYPE:
STRING
DEFAULT:
S_SQUARED
OPTIONS:
S_SQUARED
Compute first order exchange in the single-exchange (“
") approximation.
S_INVERSE
Compute the exact first order exchange.
RECOMMENDATION:
The single-exchange approximation is expected to be adequate except possibly at very short intermolecular distances, and is somewhat faster to compute.
SAPT_ORDER
Selects the order in perturbation theory for a SAPT calculation.
TYPE:
STRING
DEFAULT:
SAPT2
OPTIONS:
SAPT1
First order SAPT.
SAPT2
Second order SAPT.
ELST
First-order Rayleigh-Schrödinger perturbation theory.
RSPT
Second-order Rayleigh-Schrödinger perturbation theory.
RECOMMENDATION:
SAPT2 is the most meaningful.
SAPT_PRINT
Controls level of printing in SAPT.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
Integer print level
RECOMMENDATION:
Larger values generate additional output.
SAPT
Requests a SAPT calculation.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Run a SAPT calculation.
FALSE
Do not run SAPT.
RECOMMENDATION:
If SAPT is set to TRUE, one should also specify XPOL = TRUE and XPOL_MPOL_ORDER = GAS.
SASF_RPA
Do an SA-SF-CIS/DFT calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not do an SA-SF-CIS/DFT calculation.
TRUE
Do an SA-SF-CIS/DFT calculation (requires ROHF ground state).
RECOMMENDATION:
None
SAVE_LAST_GPX
Save the last
when calculating dynamic polarizabilities in order to call the MOProp code in a second run, via MOPROP = 102.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
False
1
True
RECOMMENDATION:
None
SCALE_NUCLEAR_CHARGE
Scales charge of each nuclei by a certain value. The nuclear repulsion energy is calculated for the unscaled nuclear charges.
TYPE:
INTEGER
DEFAULT:
0
No scaling.
OPTIONS:
A total positive charge of (1+
RECOMMENDATION:
NONE
SCFMI_FREEZE_SS
Keep the first several fragments unrelaxed in an SCFMI calculation.
TYPE:
INTEGER
DEFAULT:
0 (all fragments are active)
OPTIONS:
Freeze the first
RECOMMENDATION:
None
SCFMI_MODE
Determine whether generalized SCFMI is used and also the property of the working basis.
TYPE:
INTEGER
DEFAULT:
0 (“1" is used by basic “EDA2" calculations).
OPTIONS:
0
AO-block based SCFMI (the original definition of ALMOs).
1
Generalized SCFMI with basis vectors that are non-orthogonal between fragments.
2
Generalized SCFMI with basis vectors that are orthogonal between fragments.
RECOMMENDATION:
None
SCF_ALGORITHM
Algorithm used for converging the SCF.
TYPE:
STRING
DEFAULT:
DIIS
Pulay DIIS.
OPTIONS:
DIIS
Pulay DIIS.
DM
Direct minimizer.
DIIS_DM
Uses DIIS initially, switching to direct minimizer for later iterations
(See THRESH_DIIS_SWITCH, MAX_DIIS_CYCLES).
DIIS_GDM
Use DIIS and then later switch to geometric direct minimization
(See THRESH_DIIS_SWITCH, MAX_DIIS_CYCLES).
GDM
Geometric Direct Minimization.
RCA
Relaxed constraint algorithm
RCA_DIIS
Use RCA initially, switching to DIIS for later iterations (see
THRESH_RCA_SWITCH and MAX_RCA_CYCLES described
later in this chapter)
ROOTHAAN
Roothaan repeated diagonalization.
RECOMMENDATION:
Use DIIS unless performing a restricted open-shell calculation, in which case GDM is recommended. If DIIS fails to find a reasonable approximate solution in the initial iterations, RCA_DIIS is the recommended fallback option. If DIIS approaches the correct solution but fails to finally converge, DIIS_GDM is the recommended fallback.
SCF_CONVERGENCE
SCF is considered converged when the wave function error is less that
. Adjust the value of THRESH at the same time. (Starting with Q-Chem 3.0, the DIIS error is measured by the maximum error rather than the RMS error as in earlier versions.)
TYPE:
INTEGER
DEFAULT:
5
For single point energy calculations.
8
For geometry optimizations and vibrational analysis.
8
For SSG calculations, see Chapter 6.
OPTIONS:
User-defined
RECOMMENDATION:
Tighter criteria for geometry optimization and vibration analysis. Larger values provide more significant figures, at greater computational cost.
SCF_FINAL_PRINT
Controls level of output from SCF procedure to Q-Chem output file at the end of the SCF.
TYPE:
INTEGER
DEFAULT:
0
No extra print out.
OPTIONS:
0
No extra print out.
1
Orbital energies and break-down of SCF energy.
2
Level 1 plus MOs and density matrices.
3
Level 2 plus Fock and density matrices.
RECOMMENDATION:
The break-down of energies is often useful (level 1).
SCF_GUESS_ALWAYS
Switch to force the regeneration of a new initial guess for each series of SCF iterations (for use in geometry optimization).
TYPE:
LOGICAL
DEFAULT:
False
OPTIONS:
False
Do not generate a new guess for each series of SCF iterations in an
optimization; use MOs from the previous SCF calculation for the guess,
if available.
True
Generate a new guess for each series of SCF iterations in a geometry
optimization.
RECOMMENDATION:
Use the default unless SCF convergence issues arise
SCF_GUESS_MIX
Controls mixing of LUMO and HOMO to break symmetry in the initial guess. For unrestricted jobs, the mixing is performed only for the alpha orbitals.
TYPE:
INTEGER
DEFAULT:
0 (FALSE)
Do not mix HOMO and LUMO in SCF guess.
OPTIONS:
0 (FALSE)
Do not mix HOMO and LUMO in SCF guess.
1 (TRUE)
Add 10% of LUMO to HOMO to break symmetry.
Add
of LUMO to HOMO (
).
RECOMMENDATION:
When performing unrestricted calculations on molecules with an even number of electrons, it is often necessary to break alpha/beta symmetry in the initial guess with this option, or by specifying input for $occupied.
SCF_GUESS_PRINT
Controls printing of guess MOs, Fock and density matrices.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not print guesses.
SAD
1
Atomic density matrices and molecular matrix.
2
Level 1 plus density matrices.
CORE and GWH
1
No extra output.
2
Level 1 plus Fock and density matrices and, MO coefficients and
eigenvalues.
READ
1
No extra output
2
Level 1 plus density matrices, MO coefficients and eigenvalues.
RECOMMENDATION:
None
SCF_GUESS
Specifies the initial guess procedure to use for the SCF.
TYPE:
STRING
DEFAULT:
SAD
Superposition of atomic densities (available only with standard basis sets)
GWH
For ROHF where a set of orbitals are required.
FRAGMO
For a fragment MO calculation
OPTIONS:
CORE
Diagonalize core Hamiltonian
SAD
Superposition of atomic density
SADMO
Purified superposition of atomic densities (available only with standard basis sets)
GWH
Apply generalized Wolfsberg-Helmholtz approximation
READ
Read previous MOs from disk
FRAGMO
Superimposing converged fragment MOs
RECOMMENDATION:
SAD or SADMO guess for standard basis sets. For general basis sets, it is best to use the BASIS2 $rem. Alternatively, try the GWH or core Hamiltonian guess. For ROHF it can be useful to READ guesses from an SCF calculation on the corresponding cation or anion. Note that because the density is made spherical, this may favor an undesired state for atomic systems, especially transition metals. Use FRAGMO in a fragment MO calculation.
SCF_MINFIND_INCREASEFACTOR
Controls how the height of the penalty function changes when repeatedly trapped at the same solution
TYPE:
INTEGER
DEFAULT:
10100 meaning 1.01
OPTIONS:
corresponding to
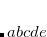
RECOMMENDATION:
If the algorithm converges to a solution which corresponds to a previously located solution, increase both the normalization N and the width lambda of the penalty function there. Then do a restart.
SCF_MINFIND_INITLAMBDA
Control the initial width of the penalty function.
TYPE:
INTEGER
DEFAULT:
02000 meaning 2.000
OPTIONS:
corresponding to
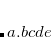
RECOMMENDATION:
The initial inverse-width (i.e., the inverse-variance) of the Gaussian to place to fill solution’s well. Measured in electrons
. Increasing this will repeatedly converging on the same solution.
SCF_MINFIND_INITNORM
Control the initial height of the penalty function.
TYPE:
INTEGER
DEFAULT:
01000 meaning 1.000
OPTIONS:
RECOMMENDATION:
The initial normalization of the Gaussian to place to fill a well. Measured in hartrees.
SCF_MINFIND_MIXENERGY
Specify the active energy range when doing Active mixing
TYPE:
INTEGER
DEFAULT:
00200 meaning 00.200
OPTIONS:
RECOMMENDATION:
The standard deviation of the Gaussian distribution used to select the orbitals for mixing (centered on the Fermi level). Measured in Hartree. To find less-excited solutions, decrease this value
SCF_MINFIND_MIXMETHOD
Specify how to select orbitals for random mixing
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Random mixing: select from any orbital to any orbital.
1
Active mixing: select based on energy, decaying with distance from the Fermi level.
2
Active Alpha space mixing: select based on energy, decaying with distance from the
Fermi level only in the alpha space.
RECOMMENDATION:
Random mixing will often find very high energy solutions. If lower energy solutions are desired, use 1 or 2.
SCF_MINFIND_NRANDOMMIXES
Control how many random mixes to do to generate new orbitals
TYPE:
INTEGER
DEFAULT:
10
OPTIONS:
Perform
RECOMMENDATION:
This is the number of occupied/virtual pairs to attempt to mix, per separate density (i.e., for unrestricted calculations both alpha and beta space will get this many rotations). If this is negative then only mix the highest 25% occupied and lowest 25% virtuals.
SCF_MINFIND_RANDOMMIXING
Control how to choose new orbitals after locating a solution
TYPE:
INTEGER
DEFAULT:
00200 meaning .02 radians
OPTIONS:
RECOMMENDATION:
After locating an SCF solution, the orbitals are mixed randomly to move to a new position in orbital space. For each occupied and virtual orbital pair picked at random and rotate between them by a random angle between 0 and this. If this is negative then use exactly this number, e.g.,
will almost exactly swap orbitals. Any number
will cause the orbitals to be swapped exactly.
SCF_MINFIND_READDISTTHRESH
The distance threshold at which to consider two solutions the same
TYPE:
INTEGER
DEFAULT:
00100 meaning 0.1
OPTIONS:
RECOMMENDATION:
The threshold to regard a minimum as the same as a read in minimum. Measured in electrons. If two minima are closer together than this, reduce the threshold to distinguish them.
SCF_MINFIND_RESTARTSTEPS
Restart with new orbitals if no minima have been found within this many steps
TYPE:
INTEGER
DEFAULT:
300
OPTIONS:
Restart after
RECOMMENDATION:
If the SCF calculation spends many steps not finding a solution, lowering this number may speed up solution-finding. If the system converges to solutions very slowly, then this number may need to be raised.
SCF_MINFIND_RUNCORR
Run post-SCF correlated methods on multiple SCF solutions
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
If this is set
, then run correlation methods for all found SCF solutions.
RECOMMENDATION:
Post-HF correlation methods should function correctly with excited SCF solutions, but their convergence is often much more difficult owing to intruder states.
SCF_MINFIND_WELLTHRESH
Specify what SCF_MINFIND believes is the basin of a solution
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
RECOMMENDATION:
When the DIIS error is less than
SCF_PRINT_FRGM
Controls the output of Q-Chem jobs on isolated fragments.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
The output is printed to the parent job output file.
FALSE
The output is not printed.
RECOMMENDATION:
Use TRUE if details about isolated fragments are important.
SCF_PRINT
Controls level of output from SCF procedure to Q-Chem output file.
TYPE:
INTEGER
DEFAULT:
0
Minimal, concise, useful and necessary output.
OPTIONS:
0
Minimal, concise, useful and necessary output.
1
Level 0 plus component breakdown of SCF electronic energy.
2
Level 1 plus density, Fock and MO matrices on each cycle.
3
Level 2 plus two-electron Fock matrix components (Coulomb, HF exchange
and DFT exchange-correlation matrices) on each cycle.
RECOMMENDATION:
Proceed with care; can result in extremely large output files at level 2 or higher. These levels are primarily for program debugging.
SCF_READMINIMA
Read in solutions from a previous SCF meta-dynamics calculation
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Read in
Read in
RECOMMENDATION:
This may not actually locate all solutions required and will probably locate others too. The SCF will also stop when the number of solutions specified in SCF_SAVEMINIMA are found. Solutions from other geometries may also be read in and used as starting orbitals. If a solution is found and matches one that is read in within SCF_MINFIND_READDISTTHRESH, its orbitals are saved in that position for any future calculations. The algorithm works by restarting from the orbitals and density of a the minimum it is attempting to find. After 10 failed restarts (defined by SCF_MINFIND_RESTARTSTEPS), it moves to another previous minimum and attempts to locate that instead. If there are no minima to find, the restart does random mixing (with 10 times the normal random mixing parameter).
SCF_SAVEMINIMA
Turn on SCF meta-dynamics and specify how many solutions to locate.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not use SCF meta-dynamics
Attempt to find
RECOMMENDATION:
Perform SCF Orbital meta-dynamics and attempt to locate
SET_QUADRATIC
Determines whether to include full quadratic response contributions for TDDFT.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Include full quadratic response contributions for TDDFT.
FALSE
Use pseudo-wave function approach.
RECOMMENDATION:
The pseudo-wave function approach is usually accurate enough and is free of accidental singularities. Consult Refs. Zhang:2015a and Ou:2015 for additional guidance.
SET_STATE_DERIV
Sets the excited state index for analytical gradient calculation for geometry optimizations and vibrational analysis with SOS-CIS(D
)
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Select the
RECOMMENDATION:
Check to see that the states do no change order during an optimization. For closed-shell systems, either CIS_SINGLETS or CIS_TRIPLETS must be set to false.
SFX_AMP_OCC_A
Defines a customer amplitude guess vector in SF-XCIS method.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
builds a guess amplitude with an
RECOMMENDATION:
Only use when default guess is not satisfactory.
SFX_AMP_VIR_B
Defines a user-specified amplitude guess vector in SF-XCIS method.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
builds a guess amplitude with a
RECOMMENDATION:
Only use when default guess is not satisfactory.
SF_STATES
Controls the number of excited spin-flip states to calculate.
TYPE:
INTEGER
DEFAULT:
0
Do not perform a SF-ADC calculation
OPTIONS:
Number of states to calculate for each irrep or
Compute
RECOMMENDATION:
Use this variable to define the number of excited states in the case of a spin-flip calculation. SF-ADC is available for ADC(2)-s, ADC(2)-x and ADC(3).
SKIP_CHARGE_SELF_INTERACT
Ignores the electrostatic interactions among external charges in a QM/MM calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
No electrostatic interactions among external charges.
FALSE
Computes the electrostatic interactions among external charges.
RECOMMENDATION:
None
SKIP_CIS_RPA
Skips the solution of the CIS, RPA, TDA or TDDFT equations for wave function analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE / FALSE
RECOMMENDATION:
Set to true to speed up the generation of plot data if the same calculation has been run previously with the scratch files saved.
SOLVENT_METHOD
Sets the preferred solvent method.
TYPE:
STRING
DEFAULT:
0
OPTIONS:
0
Do not use a solvation model.
ONSAGER
Use the Kirkwood-Onsager model (Section 12.2.1).
PCM
Use an apparent surface charge, polarizable continuum model
(Section 12.2.2).
ISOSVP
Use the isodensity implementation of the SS(V)PE model
(Section 12.2.5).
COSMO
Use COSMO (similar to C-PCM but with an outlying charge
correction;[Klamt and Jonas(1996), Baldridge and Klamt(1997)] see Section 12.2.7).
SM8
Use version 8 of the Cramer-Truhlar SM
model (Section 12.2.8.1).
SM12
Use version 12 of the SM
SMD
Use SMD (Section 12.2.8.3).
CHEM_SOL
Use the Langevin Dipoles model (Section 12.2.9).
RECOMMENDATION:
Consult the literature. PCM is a collective name for a family of models and additional input options may be required in this case, in order to fully specify the model. (See Section 12.2.2.) Several versions of SM12 are available as well, as discussed in Section 12.2.8.2.
SOLVE_PEQ
Perform a solvation free energy calculation on a Cartesian grid using Poisson equation boundary conditions.
TYPE:
STRING
DEFAULT:
False
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
None.
SOS_FACTOR
Sets the scaling parameter
TYPE:
INTEGER
DEFAULT:
1300000
corresponding to 1.30
OPTIONS:

RECOMMENDATION:
Use the default
SOS_UFACTOR
Sets the scaling parameter
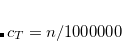
TYPE:
INTEGER
DEFAULT:
151
For SOS-CIS(D), corresponding to 1.51
140
For SOS-CIS(D
OPTIONS:
RECOMMENDATION:
Use the default
SPIN_FLIP_XCIS
Do a SF-XCIS calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not do an SF-XCIS calculation.
TRUE
Do an SF-XCIS calculation (requires ROHF triplet ground state).
RECOMMENDATION:
None
SPIN_FLIP
Selects whether to perform a standard excited state calculation, or a spin-flip calculation. Spin multiplicity should be set to 3 for systems with an even number of electrons, and 4 for systems with an odd number of electrons.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
None
SRC_DFT
Selects form of the short-range corrected functional.
TYPE:
INTEGER
DEFAULT:
No default
OPTIONS:
1
SRC1 functional.
2
SRC2 functional.
RECOMMENDATION:
None
SSG
Controls the calculation of the SSG wave function.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not compute the SSG wave function
1
Do compute the SSG wave function
RECOMMENDATION:
See also the UNRESTRICTED and DIIS_SUBSPACE_SIZE $rem variables.
SSS_FACTOR
Controls the strength of the same-spin component of PT2 correlation energy.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Corresponding to
in Eq. (5.60).
RECOMMENDATION:
NONE
STABILITY_ANALYSIS
Performs stability analysis for a HF or DFT solution.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform stability analysis.
FALSE
Do not perform stability analysis.
RECOMMENDATION:
Set to TRUE when a HF or DFT solution is suspected to be unstable.
STATE_ANALYSIS
Controls the analysis and export of excited state densities and orbitals (see 11.2.6 for details).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform excited state analyses.
FALSE
No excited state analyses or export will be performed.
RECOMMENDATION:
Set to TRUE, if detailed analysis of the excited states is required or if density or orbital plots are needed.
STATE_FOLLOW
Turns on state following.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not use state-following.
TRUE
Use state-following.
RECOMMENDATION:
None.
STS_ACCEPTOR
Define the acceptor molecular fragment.
TYPE:
STRING
DEFAULT:
0
No acceptor fragment is defined.
OPTIONS:
Acceptor fragment is in the
RECOMMENDATION:
Note no space between the hyphen and the numbers
STS_DONOR
Define the donor fragment.
TYPE:
STRING
DEFAULT:
0
No donor fragment is defined.
OPTIONS:
Donor fragment is in the
RECOMMENDATION:
Note no space between the hyphen and the numbers
STS_FCD
Control the calculation of FCD for ET couplings.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform an FCD calculation.
TRUE
Include an FCD calculation.
RECOMMENDATION:
None
STS_FED
Control the calculation of FED for EET couplings.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform a FED calculation.
TRUE
Include a FED calculation.
RECOMMENDATION:
None
STS_FSD
Control the calculation of FSD for EET couplings.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform a FSD calculation.
TRUE
Include a FSD calculation.
RECOMMENDATION:
For RCIS triplets, FSD and FED are equivalent. FSD will be automatically switched off and perform a FED calculation.
STS_GMH
Control the calculation of GMH for ET couplings.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform a GMH calculation.
TRUE
Include a GMH calculation.
RECOMMENDATION:
When set to true computes Mulliken-Hush electronic couplings. It yields the generalized Mulliken-Hush couplings as well as the transition dipole moments for each pair of excited states and for each excited state with the ground state.
STS_MOM
Control calculation of the transition moments between excited states in the CIS and TDDFT calculations (including SF variants).
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not calculate state-to-state transition moments.
TRUE
Do calculate state-to-state transition moments.
RECOMMENDATION:
When set to true requests the state-to-state dipole transition moments for all pairs of excited states and for each excited state with the ground state.
SVP_CAVITY_CONV
Determines the convergence value of the iterative isodensity cavity procedure.
TYPE:
INTEGER
DEFAULT:
10
OPTIONS:
Convergence threshold set to
RECOMMENDATION:
The default value unless convergence problems arise.
SVP_CHARGE_CONV
Determines the convergence value for the charges on the cavity. When the change in charges fall below this value, if the electron density is converged, then the calculation is considered converged.
TYPE:
INTEGER
DEFAULT:
7
OPTIONS:
Convergence threshold set to
RECOMMENDATION:
The default value unless convergence problems arise.
SVP_GUESS
Specifies how and if the solvation module will use a given guess for the charges and cavity points.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No guessing.
1
Read a guess from a previous Q-Chem solvation computation.
2
Use a guess specified by the $svpirf section from the input
RECOMMENDATION:
It is helpful to also set SCF_GUESS to READ when using a guess from a previous Q-Chem run.
SVP_MEMORY
Specifies the amount of memory for use by the solvation module.
TYPE:
INTEGER
DEFAULT:
125
OPTIONS:
RECOMMENDATION:
The default should be fine for medium size molecules with the default Lebedev grid, only increase if needed.
SVP_PATH
Specifies whether to run a gas phase computation prior to performing the solvation procedure.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
runs a gas-phase calculation and after
convergence runs the SS(V)PE computation.
1
does not run a gas-phase calculation.
RECOMMENDATION:
Running the gas-phase calculation provides a good guess to start the solvation stage and provides a more complete set of solvated properties.
SYMMETRY_DECOMPOSITION
Determines symmetry decompositions to calculate.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
0
No symmetry decomposition.
1
Calculate MO eigenvalues and symmetry (if available).
2
Perform symmetry decomposition of kinetic energy and nuclear attraction
matrices.
RECOMMENDATION:
None
SYMMETRY
Controls the efficiency through the use of point group symmetry for calculating integrals.
TYPE:
LOGICAL
DEFAULT:
TRUE
Use symmetry for computing integrals.
OPTIONS:
TRUE
Use symmetry when available.
FALSE
Do not use symmetry. This is always the case for RIMP2 jobs
RECOMMENDATION:
Use the default unless benchmarking. Note that symmetry usage is disabled for RIMP2, FFT, and QM/MM jobs.
SYM_IGNORE
Controls whether or not Q-Chem determines the point group of the molecule and reorients the molecule to the standard orientation.
TYPE:
LOGICAL
DEFAULT:
FALSE
Do determine the point group (disabled for RIMP2 jobs).
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Use the default unless you do not want the molecule to be reoriented. Note that symmetry usage is disabled for RIMP2 jobs.
SYM_TOL
Controls the tolerance for determining point group symmetry. Differences in atom locations less than
are treated as zero.
TYPE:
INTEGER
DEFAULT:
5
Corresponding to
OPTIONS:
User defined.
RECOMMENDATION:
Use the default unless the molecule has high symmetry which is not being correctly identified. Note that relaxing this tolerance too much may introduce errors into the calculation.
TAO_DFT_THETA_NDP
Controls the value of the fictitious temperature
in TAO-DFT.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
(hartrees), where
is the value of TAO_DFT_THETA
RECOMMENDATION:
NONE
TAO_DFT_THETA
Controls the value of the fictitious temperature
TYPE:
INTEGER
DEFAULT:
7
OPTIONS:
RECOMMENDATION:
NONE
TAO_DFT
Controls whether to use TAO-DFT.
TYPE:
Boolean
DEFAULT:
false
OPTIONS:
false
Do not use TAO-DFT
true
Use TAO-DFT
RECOMMENDATION:
NONE
TDDFT_MI
Perform an TDDFT(MI) calculation
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not perform an TDDFT(MI) calculation
TRUE
Perform an TDDFT(MI) calculation
RECOMMENDATION:
False
THRESH_DIIS_SWITCH
The threshold for switching between DIIS extrapolation and direct minimization of the SCF energy is
when SCF_ALGORITHM is DIIS_GDM or DIIS_DM. See also MAX_DIIS_CYCLES
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
User-defined.
RECOMMENDATION:
None
THRESH_RCA_SWITCH
The threshold for switching between RCA and DIIS when SCF_ALGORITHM is RCA_DIIS.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
N
Algorithm changes from RCA to DIIS when Error is less than
RECOMMENDATION:
None
THRESH
Cutoff for neglect of two electron integrals.
).
TYPE:
INTEGER
DEFAULT:
8
For single point energies.
10
For optimizations and frequency calculations.
14
For coupled-cluster calculations.
OPTIONS:
for a threshold of
RECOMMENDATION:
Should be at least three greater than SCF_CONVERGENCE. Increase for more significant figures, at greater computational cost.
TIME_STEP
Specifies the molecular dynamics time step, in atomic units (1 a.u. = 0.0242 fs).
TYPE:
INTEGER
DEFAULT:
None.
OPTIONS:
User-specified.
RECOMMENDATION:
Smaller time steps lead to better energy conservation; too large a time step may cause the job to fail entirely. Make the time step as large as possible, consistent with tolerable energy conservation.
TRANS_ENABLE
To invoke the molecular transport code.
TYPE:
INTEGER
DEFAULT:
0
Do not perform transport calculations (default).
OPTIONS:
1
Perform transport calculations.
Print matrices needed for generating bulk model files.
RECOMMENDATION:
None
TRNSS
Controls whether reduced single excitation space is used.
TYPE:
LOGICAL
DEFAULT:
FALSE
Use full excitation space.
OPTIONS:
TRUE
Use reduced excitation space.
RECOMMENDATION:
None
TRTYPE
Controls how reduced subspace is specified.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Select orbitals localized on a set of atoms.
2
Specify a set of orbitals.
3
Specify a set of occupied orbitals, include excitations to all virtual orbitals.
RECOMMENDATION:
None
TSVDW
Flag to switch on the TS-vdW method
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not apply TS-vdW.
1
Apply the TS-vdW method to obtain the TS-vdW energy.
2
Apply the TS-vdW method to obtain the TS-vdW energy and corresponding gradients.
RECOMMENDATION:
Since TS-vdW is itself a form of dispersion correction, it should not be used in conjunction with any of the dispersion corrections described in Section 5.7.2.
UNRESTRICTED
Controls the use of restricted or unrestricted orbitals.
TYPE:
LOGICAL
DEFAULT:
FALSE
Closed-shell systems.
TRUE
Open-shell systems.
OPTIONS:
FALSE
Constrain the spatial part of the alpha and beta orbitals to be the same.
TRUE
Do not Constrain the spatial part of the alpha and beta orbitals.
RECOMMENDATION:
Use the default unless ROHF is desired. Note that for unrestricted calculations on systems with an even number of electrons it is usually necessary to break
USECUBLAS_THRESH
Sets threshold of matrix size sent to GPU (smaller size not worth sending to GPU).
TYPE:
INTEGER
DEFAULT:
250
OPTIONS:
n
user-defined threshold
RECOMMENDATION:
Use the default value. Anything less can seriously hinder the GPU acceleration
USER_CONNECT
Enables explicitly defined bonds.
TYPE:
STRING
DEFAULT:
FALSE
OPTIONS:
TRUE
Bond connectivity is read from the $molecule section
FALSE
Bond connectivity is determined by atom proximity
RECOMMENDATION:
Set to TRUE if bond connectivity is known, in which case this connectivity must be specified in the $molecule section. This greatly accelerates MM calculations.
USE_LIBPT
Enable libpt for CCSD(T) calculations in CCMAN2.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
FALSE
RECOMMENDATION:
libpt is now used by default in all real-valued CC/EOM-CC calculations
USE_MGEMM
Use the mixed-precision matrix scheme (MGEMM) if you want to make calculations in your card in single-precision (or if you have a single-precision-only GPU), but leave some parts of the RI-MP2 calculation in double precision)
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
MGEMM disabled
TRUE
MGEMM enabled
RECOMMENDATION:
Use when having single-precision cards
USE_RVV10
Used to turn on the rVV10 NLC functional
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Use VV10 NLC (the default for NL_CORRELATION)
TRUE
Use rVV10 NLC
RECOMMENDATION:
Set to TRUE if the rVV10 NLC is desired.
VARTHRESH
Controls the temporary integral cut-off threshold.
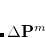
TYPE:
INTEGER
DEFAULT:
0
Turns VARTHRESH off
OPTIONS:
User-defined threshold
RECOMMENDATION:
3 has been found to be a practical level, and can slightly speed up SCF evaluation.
VCI
Specifies the number of quanta involved in the VCI calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined. Maximum value is 10.
RECOMMENDATION:
The availability depends on the memory of the machine. Memory allocation for VCI calculation is the square of
with double precision. For example, a machine with 1.5 Gb memory and for molecules with fewer than 4 atoms, VCI(10) can be carried out, for molecule containing fewer than 5 atoms, VCI(6) can be carried out, for molecule containing fewer than 6 atoms, VCI(5) can be carried out. For molecules containing fewer than 50 atoms, VCI(2) is available. VCI(1) and VCI(3) usually overestimated the true energy while VCI(4) usually gives an answer close to the converged energy.
VIBMAN_PRINT
Controls level of extra print out for vibrational analysis.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Standard full information print out.
If VCI is TRUE, overtones and combination bands are also printed.
3
Level 1 plus vibrational frequencies in atomic units.
4
Level 3 plus mass-weighted Hessian matrix, projected mass-weighted Hessian
matrix.
6
Level 4 plus vectors for translations and rotations projection matrix.
RECOMMENDATION:
Use the default.
WANG_ZIEGLER_KERNEL
Controls whether to use the Wang-Ziegler non-collinear exchange-correlation kernel in a SF-DFT calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not use non-collinear kernel.
TRUE
Use non-collinear kernel.
RECOMMENDATION:
None
WAVEFUNCTION_ANALYSIS
Controls the running of the default wave function analysis tasks.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Perform default wave function analysis.
FALSE
Do not perform default wave function analysis.
RECOMMENDATION:
None
WIG_GRID
Specify angular Lebedev grid for Wigner intracule calculations.
TYPE:
INTEGER
DEFAULT:
194
OPTIONS:
Lebedev grids up to 5810 points.
RECOMMENDATION:
Larger grids if high accuracy required.
WIG_LEB
Use Lebedev quadrature to evaluate Wigner integrals.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Evaluate Wigner integrals through series summation.
TRUE
Use quadrature for Wigner integrals.
RECOMMENDATION:
None
WIG_MEM
Reduce memory required in the evaluation of
.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not use low memory option.
TRUE
Use low memory option.
RECOMMENDATION:
The low memory option is slower, so use the default unless memory is limited.
WRITE_WFN
Specifies whether or not a wfn file is created, which is suitable for use with AIMPAC. Note that the output to this file is currently limited to
orbitals, which is the highest angular momentum implemented in AIMPAC.
TYPE:
STRING
DEFAULT:
(NULL)
No output file is created.
OPTIONS:
filename
Specifies the output file name. The suffix .wfn will
be appended to this name.
RECOMMENDATION:
None
XCIS
Do an XCIS calculation in addition to a CIS calculation.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not do an XCIS calculation.
TRUE
Do an XCIS calculation (requires ROHF ground state).
RECOMMENDATION:
None
XC_GRID
Specifies the type of grid to use for DFT calculations.
TYPE:
INTEGER
DEFAULT:
Functional-dependent; see Table 5.3.
OPTIONS:
0
Use SG-0 for H, C, N, and O; SG-1 for all other atoms.
Use SG-
, or 3
A string of two six-digit integers
and
, where
and
points, which must be an allowed value from Table 5.2 in Section 5.5.
Similar format for Gauss-Legendre grids, with the six-digit integer
to the number of radial points and the six-digit integer
Gauss-Legendre angular points,
.
RECOMMENDATION:
Use the default unless numerical integration problems arise. Larger grids may be required for optimization and frequency calculations.
XC_SMART_GRID
Uses SG-0 (where available) for early SCF cycles, and switches to the (larger) target grid specified by XC_GRID for final cycles of the SCF.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE (or 1)
Use the smaller grid for the initial cycles.
FALSE (or 0)
Use the target grid for all SCF cycles.
RECOMMENDATION:
The use of the smart grid can save some time on initial SCF cycles.
XOPT_SEAM_ONLY
Orders an intersection seam search only, no minimization is to perform.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Find a point on the intersection seam and stop.
FALSE
Perform a minimization of the intersection seam.
RECOMMENDATION:
In systems with a large number of degrees of freedom it might be useful to locate the seam first setting this option to TRUE and use that geometry as a starting point for the minimization.
XOPT_STATE_1, XOPT_STATE_2
Specify two electronic states the intersection of which will be searched.
TYPE:
[INTEGER, INTEGER, INTEGER]
DEFAULT:
No default value (the option must be specified to run this calculation)
OPTIONS:
[spin, irrep, state]
spin = 0
Addresses states with low spin,
see also EE_SINGLETS or IP_STATES,EA_STATES.
spin = 1
Addresses states with high spin,
see also EE_TRIPLETS.
irrep
Specifies the irreducible representation to which
the state belongs, for
point group symmetry
irrep = 1 for
, irrep = 2 for
,
irrep = 3 for
, irrep = 4 for
.
state
Specifies the state number within the irreducible
representation, state = 1 means the lowest excited
state, state = 2 is the second excited state, etc..
0, 0, -1
Ground state.
RECOMMENDATION:
Only intersections of states with different spin or symmetry can be calculated at this time.
XPOL_CHARGE_TYPE
Controls the type of atom-centered embedding charges for XPol calculations.
TYPE:
STRING
DEFAULT:
QLOWDIN
OPTIONS:
QLOWDIN
Löwdin charges.
QMULLIKEN
Mulliken charges.
QCHELPG
ChElPG charges.
RECOMMENDATION:
Problems with Mulliken charges in extended basis sets can lead to XPol convergence failure. Löwdin charges tend to be more stable, and ChElPG charges are both robust and provide an accurate electrostatic embedding. However, ChElPG charges are more expensive to compute, and analytic energy gradients are not yet available for this choice.
XPOL_MPOL_ORDER
Controls the order of multipole expansion that describes electrostatic interactions.
TYPE:
STRING
DEFAULT:
CHARGES
OPTIONS:
GAS
No electrostatic embedding; monomers are in the gas phase.
CHARGES
Charge embedding.
DENSITY
Density embedding.
RECOMMENDATION:
Should be set to GAS to do a dimer SAPT calculation (see Section 13.11).
XPOL_OMEGA
Controls the range-separation parameter,
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Use different
FALSE
Use a single value of
RECOMMENDATION:
If FALSE, the $rem variable OMEGA should be used to specify the single value of
OMEGA/1000, in atomic units.
XPOL_PRINT
Print level for XPol calculations.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
Integer print level
RECOMMENDATION:
Higher values prints more information
XPOL
Perform a self-consistent XPol calculation.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform an XPol calculation.
FALSE
Do not perform an XPol calculation.
RECOMMENDATION:
NONE
Z_EXTRAP_ORDER
Specifies the polynomial order
TYPE:
INTEGER
DEFAULT:
0
Do not perform
OPTIONS:
Extrapolate using an
RECOMMENDATION:
None
Z_EXTRAP_POINTS
Specifies the number
TYPE:
INTEGER
DEFAULT:
0
Do not perform response equation extrapolation.
OPTIONS:
Save

RECOMMENDATION:
Using the default
extrapolation was shown to provide the greatest speedup. At this setting, a 2–3-fold reduction in iterations was demonstrated.