4.3 Basic SCF Job Control
4.3.1 Overview
As of version 5.1, Q-Chem uses a new SCF package, GEN_SCFMAN, developed by E. J. Sundstrom, P. R. Horn and many other coworkers. In addition to supporting the basic features of the previous SCF package (e.g. restricted, unrestricted and restricted open-shell HF/KS-DFT calculations), many new features are now available in Q-Chem, including:
Addition of several useful SCF convergence algorithms and support for user-specified hybrid algorithm (Sect. 4.5.8).
More general and user-friendly internal stability analysis and automatic correction for the energy minimum (Sect. 4.5.9).
GEN_SCFMAN also supports a wider range of orbital types, including complex orbitals. A full list of supported orbitals is:
Restricted (R): typically appropriate for closed shell molecules at their equilibrium geometry, where electrons occupy orbitals in pairs.
Unrestricted (U): - appropriate for radicals with an odd number of electrons, and also for molecules with even numbers of electrons where not all electrons are paired, e.g., stretched bonds and diradicals.
Restricted open-shell (RO): for open-shell molecules, where the
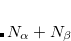 and
and  orbitals are constrained to be identical.
orbitals are constrained to be identical. Open-shell singlet ROSCF (OS_RO): see the “ROKS" method documented in Section 7.5.
Generalized (G): i.e., each MO is associated with both
and spin components. The use of complex orbitals (with Hartree-Fock only): restricted (CR), unrestricted (CU), and generalized (CG).
Aspects of an SCF calculation such as the SCF guess, the use of efficient algorithms to construct the Fock matrix like occ-RI-K (see Section 4.6.9), are unaffected by the use of GEN_SCFMAN. Likewise, using GEN_SCFMAN does not make any difference to the post-SCF procedures such as correlated methods, excited state calculations and evaluation of molecular properties.
It should be noted that many special features (e.g. dual-basis SCF, CDFT, etc.) based on Q-Chem’s old SCF code are not yet supported in GEN_SCFMAN. They will become available in the future.
4.3.1.1 Job Control
The following two $rem variables must be specified in order to run HF calculations:
METHOD
Specifies the exchange-correlation functional.
TYPE:
STRING
DEFAULT:
No default
OPTIONS:
NAME
Use METHOD = NAME, where NAME is either HF for Hartree-Fock theory or
else one of the DFT methods listed in Section 5.3.4.
RECOMMENDATION:
In general, consult the literature to guide your selection. Our recommendations for DFT are indicated in bold in Section 5.3.4.
BASIS
Specifies the basis sets to be used.
TYPE:
STRING
DEFAULT:
No default basis set
OPTIONS:
General, Gen
User defined ($basis keyword required).
Symbol
Use standard basis sets as per Chapter 8.
Mixed
Use a mixture of basis sets (see Chapter 8).
RECOMMENDATION:
Consult literature and reviews to aid your selection.
In addition, the following $rem variables can be used to customize the SCF calculation:
GEN_SCFMAN
Use GEN_SCFMAN for the present SCF calculation.
TYPE:
BOOLEAN
DEFAULT:
TRUE
OPTIONS:
FALSE
Use the previous SCF code.
TRUE
Use GEN_SCFMAN.
RECOMMENDATION:
Set to FALSE in cases where features not yet supported by GEN_SCFMAN are needed.
PRINT_ORBITALS
Prints orbital coefficients with atom labels in analysis part of output.
TYPE:
INTEGER/LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not print any orbitals.
TRUE
Prints occupied orbitals plus 5 virtual orbitals.
NVIRT
Number of virtual orbitals to print.
RECOMMENDATION:
Use true unless more virtual orbitals are desired.
SCF_CONVERGENCE
SCF is considered converged when the wave function error is less that
. Adjust the value of THRESH at the same time. (Starting with Q-Chem 3.0, the DIIS error is measured by the maximum error rather than the RMS error as in earlier versions.)
TYPE:
INTEGER
DEFAULT:
5
For single point energy calculations.
8
For geometry optimizations and vibrational analysis.
8
For SSG calculations, see Chapter 6.
OPTIONS:
User-defined
RECOMMENDATION:
Tighter criteria for geometry optimization and vibration analysis. Larger values provide more significant figures, at greater computational cost.
UNRESTRICTED
Controls the use of restricted or unrestricted orbitals.
TYPE:
LOGICAL
DEFAULT:
FALSE
Closed-shell systems.
TRUE
Open-shell systems.
OPTIONS:
FALSE
Constrain the spatial part of the alpha and beta orbitals to be the same.
TRUE
Do not Constrain the spatial part of the alpha and beta orbitals.
RECOMMENDATION:
Use the default unless ROHF is desired. Note that for unrestricted calculations on systems with an even number of electrons it is usually necessary to break
The calculations using other more special orbital types are controlled by the following $rem variables (they are not effective if GEN_SCFMAN = FALSE):
OS_ROSCF
Run an open-shell singlet ROSCF calculation with GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
OS_ROSCF calculation is performed.
FALSE
Do not run OS_ROSCF (it will run a close-shell RSCF calculation instead).
RECOMMENDATION:
Set to TRUE if desired.
GHF
Run a generalized Hartree-Fock calculation with GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Run a GHF calculation.
FALSE
Do not use GHF.
RECOMMENDATION:
Set to TRUE if desired.
COMPLEX
Run an SCF calculation with complex MOs using GEN_SCFMAN.
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Use complex orbitals.
FALSE
Use real orbitals.
RECOMMENDATION:
Set to TRUE if desired.
COMPLEX_MIX
Mix a certain percentage of the real part of the HOMO to the imaginary part of the LUMO.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0–100
The mix angle =
COMPLEX_MIX/100.
RECOMMENDATION:
It may help find the stable complex solution (similar idea as SCF_GUESS_MIX).
Example 4.17 Restricted open-shell singlet ROSCF calculation for the first excited state of formaldehyde using GEN_SCFMAN. The first job provides the guess orbitals through a restricted SCF calculation.
$molecule
0 1
H -0.940372 0.000000 1.268098
H 0.940372 0.000000 1.268098
C 0.000000 0.000000 0.682557
O 0.000000 0.000000 -0.518752
$end
$rem
GEN_SCFMAN true
METHOD wb97x-d
BASIS def2-svpd
THRESH 14
SCF_CONVERGENCE 9
SYM_IGNORE true
$end
@@@
$molecule
read
$end
$rem
JOBTYPE sp
METHOD wb97x-d
BASIS def2-svpd
GEN_SCFMAN true
OS_ROSCF true
THRESH 14
SCF_CONVERGENCE 9
SCF_ALGORITHM diis
SYM_IGNORE true
SCF_GUESS read
$end
4.3.2 Additional Options
Listed below are a number of useful options to customize an SCF calculation. This is only a short summary of the function of these $rem variables. A full list of all SCF-related variables is provided in Appendix C. Several important sub-topics are discussed separately, including  methods for large molecules (Section 4.6), customizing the initial guess (Section 4.4), and converging the SCF calculation (Section 4.5).
methods for large molecules (Section 4.6), customizing the initial guess (Section 4.4), and converging the SCF calculation (Section 4.5).
INTEGRALS_BUFFER
Controls the size of in-core integral storage buffer.
TYPE:
INTEGER
DEFAULT:
15
15 Megabytes.
OPTIONS:
User defined size.
RECOMMENDATION:
Use the default, or consult your systems administrator for hardware limits.
DIRECT_SCF
Controls direct SCF.
TYPE:
LOGICAL
DEFAULT:
Determined by program.
OPTIONS:
TRUE
Forces direct SCF.
FALSE
Do not use direct SCF.
RECOMMENDATION:
Use the default; direct SCF switches off in-core integrals.
METECO
Sets the threshold criteria for discarding shell-pairs.
TYPE:
INTEGER
DEFAULT:
2
Discard shell-pairs below
.
OPTIONS:
1
Discard shell-pairs four orders of magnitude below machine precision.
2
Discard shell-pairs below 10
.
RECOMMENDATION:
Use the default.
THRESH
Cutoff for neglect of two electron integrals.
).
TYPE:
INTEGER
DEFAULT:
8
For single point energies.
10
For optimizations and frequency calculations.
14
For coupled-cluster calculations.
OPTIONS:

for a threshold of
.
RECOMMENDATION:
Should be at least three greater than SCF_CONVERGENCE. Increase for more significant figures, at greater computational cost.
STABILITY_ANALYSIS
Performs stability analysis for a HF or DFT solution.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Perform stability analysis.
FALSE
Do not perform stability analysis.
RECOMMENDATION:
Set to TRUE when a HF or DFT solution is suspected to be unstable.
SCF_PRINT
Controls level of output from SCF procedure to Q-Chem output file.
TYPE:
INTEGER
DEFAULT:
0
Minimal, concise, useful and necessary output.
OPTIONS:
0
Minimal, concise, useful and necessary output.
1
Level 0 plus component breakdown of SCF electronic energy.
2
Level 1 plus density, Fock and MO matrices on each cycle.
3
Level 2 plus two-electron Fock matrix components (Coulomb, HF exchange
and DFT exchange-correlation matrices) on each cycle.
RECOMMENDATION:
Proceed with care; can result in extremely large output files at level 2 or higher. These levels are primarily for program debugging.
SCF_FINAL_PRINT
Controls level of output from SCF procedure to Q-Chem output file at the end of the SCF.
TYPE:
INTEGER
DEFAULT:
0
No extra print out.
OPTIONS:
0
No extra print out.
1
Orbital energies and break-down of SCF energy.
2
Level 1 plus MOs and density matrices.
3
Level 2 plus Fock and density matrices.
RECOMMENDATION:
The break-down of energies is often useful (level 1).
4.3.3 Examples
Provided below are examples of Q-Chem input files to run ground state, HF single point energy calculations.
Example 4.18 Example Q-Chem input for a single point energy calculation on water. Note that the declaration of the single point $rem variable is redundant because it is the same as the Q-Chem default.
$molecule
0 1
O
H1 O oh
H2 O oh H1 hoh
oh = 1.2
hoh = 120.0
$end
$rem
JOBTYPE sp Single Point energy
METHOD hf Hartree-Fock
BASIS sto-3g Basis set
$end
Example 4.19 UHF/6-311G calculation on the Li atom. Note that correlation and the job type were not indicated because Q-Chem defaults automatically to no correlation and single point energies. Note also that, since the number of and electron differ, MOs default to an unrestricted formalism.
$molecule
0,2
Li
$end
$rem
METHOD HF Hartree-Fock
BASIS 6-311G Basis set
$end
Example 4.20 ROHF/6-311G calculation on the Lithium atom.
$molecule
0,2
3
$end
$rem
METHOD hf Hartree-Fock
UNRESTRICTED false Restricted MOs
BASIS 6-311G Basis set
$end
4.3.4 Symmetry
Symmetry is a powerful branch of mathematics and is often exploited in quantum chemistry, both to reduce the computational workload and to classify the final results obtained.[Takada et al.(1981)Takada, Dupuis, and King, Dupuis and King(1977), Dupuis and King(1978)] Q-Chem is able to determine the point group symmetry of the molecular nuclei and, on completion of the SCF procedure, classify the symmetry of molecular orbitals, and provide symmetry decomposition of kinetic and nuclear attraction energy (see Chapter 11).
Molecular systems possessing point group symmetry offer the possibility of large savings of computational time, by avoiding calculations of integrals which are equivalent i.e., those integrals which can be mapped on to one another under one of the symmetry operations of the molecular point group. The Q-Chem default is to use symmetry to reduce computational time, when possible.
There are several keywords that are related to symmetry, which causes frequent confusion. SYM_IGNORE controls symmetry throughout all modules. The default is FALSE. In some cases it may be desirable to turn off symmetry altogether, for example if you do not want Q-Chem to reorient the molecule into the standard nuclear orientation, or if you want to turn it off for finite difference calculations. If the SYM_IGNORE keyword is set to TRUE then the coordinates will not be altered from the input, and the point group will be set to 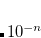 .
.
The SYMMETRY keyword controls symmetry in some integral routines. It is set to FALSE by default. Note that setting it to FALSE does not turn point group symmetry off, and does not disable symmetry in the coupled-cluster suite (CCMAN and CCMAN2), which is controlled by CC_SYMMETRY (see Chapters 6 and 7), although we noticed that sometimes it may interfere with the determination of orbital symmetries, possibly due to numerical noise. In some cases, SYMMETRY = TRUE can cause problems (poor convergence and wildly incorrect SCF energies) and turning it off can avoid these problems.
Note: The user should be aware about different conventions for defining symmetry elements. The arbitrariness affects, for example,  point group. The specific choice affects how the irreps in the affected groups are labeled. For example,
point group. The specific choice affects how the irreps in the affected groups are labeled. For example, 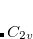 and
and 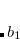 irreps in are flipped when using different conventions. Q-Chem uses non-Mulliken symmetry convention. See http://iopenshell.usc.edu/howto/symmetry for detailed explanations.
irreps in are flipped when using different conventions. Q-Chem uses non-Mulliken symmetry convention. See http://iopenshell.usc.edu/howto/symmetry for detailed explanations.
SYMMETRY
Controls the efficiency through the use of point group symmetry for calculating integrals.
TYPE:
LOGICAL
DEFAULT:
TRUE
Use symmetry for computing integrals.
OPTIONS:
TRUE
Use symmetry when available.
FALSE
Do not use symmetry. This is always the case for RIMP2 jobs
RECOMMENDATION:
Use the default unless benchmarking. Note that symmetry usage is disabled for RIMP2, FFT, and QM/MM jobs.
SYM_IGNORE
Controls whether or not Q-Chem determines the point group of the molecule and reorients the molecule to the standard orientation.
TYPE:
LOGICAL
DEFAULT:
FALSE
Do determine the point group (disabled for RIMP2 jobs).
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Use the default unless you do not want the molecule to be reoriented. Note that symmetry usage is disabled for RIMP2 jobs.
SYM_TOL
Controls the tolerance for determining point group symmetry. Differences in atom locations less than
are treated as zero.
TYPE:
INTEGER
DEFAULT:
5
Corresponding to
.
OPTIONS:
User defined.
RECOMMENDATION:
Use the default unless the molecule has high symmetry which is not being correctly identified. Note that relaxing this tolerance too much may introduce errors into the calculation.