4.1 Overview
Theoretical “model chemistries" [9] involve two principle approximations. One must specify, first of all, the type of atomic orbital (AO) basis set that will be used to construct molecular orbitals (MOs), via the “linear combination of atomic orbitals” (LCAO) ansatz, available options for which are discussed in Chapters 7 and 8. Second, one must specify the manner in which the instantaneous interactions between electrons (“electron correlation”) are to be treated. Self-consistent field (SCF) methods, in which electron correlation is described in a mean-field way, represent the simplest, most affordable, and most widely-used electronic structure methods. The SCF category of methods includes both Hartree-Fock (HF) theory as well as Kohn-Sham (KS) density functional theory (DFT). This chapter summarizes Q-Chem’s SCF capabilities, whereas Chapter 5 describes the more sophisticated (but also more computationally expensive!) post-HF, wave function-based methods for describing electron correlation. If you are new to quantum chemistry, we recommend an introductory textbook such as Refs. Hehre:1986, Szabo:1996, or Jensen:1994.
Section 4.2 provides the theoretical background behind SCF methods, including both HF and KS-DFT. In some sense, the former may be considered as a special case of the latter, and job-control $rem variables are much the same in both cases. Basic SCF job control is described in Section 4.3. Later sections introduce more specialized options that can be consulted as needed. Of particular note are the following:
Initial guesses for SCF calculations (Section 4.5). Modification of the guess is recommended in cases where the SCF calculation fails to converge.
Changing the SCF convergence algorithm (Section 4.6) is also a good strategy when the SCF calculation fails to converge.
Linear-scaling [“
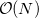 ”] and other reduced-cost methods are available for large systems (see Section 4.8).
”] and other reduced-cost methods are available for large systems (see Section 4.8). Unconventional SCF calculations. Some non-standard SCF methods with novel physical and mathematical features are available. These include:
Dual-basis SCF calculations (Section 4.9) and DFT perturbation theory (Section 4.10), which facilitate large-basis quality results but require self-consistent iterations only in a smaller basis set.
Charge-constrained DFT (CDFT; Section 4.11), in the DFT calculation is subject to the constraint that certain fragments integrate to a pre-defined numbers of electrons. This can be used to circumvent some delocalization problems in conventional DFT.
Configuration interaction (CI) with a CDFT reference state (CDFT-CI; Section 4.11.3)
SCF meta-dynamics (Section 4.12.2), which can be used to locate multiple solutions to the SCF equations and to help check that the solution obtained is actually the lowest minimum.
Some of these unconventional SCF methods are available exclusively in Q-Chem.