A.4 Delocalized Internal Coordinates
The choice of coordinate system can have a major influence on the rate of convergence during a geometry optimization. For complex potential energy surfaces with many stationary points, a different choice of coordinates can result in convergence to a different final structure.
The key attribute of a good set of coordinates for geometry optimization is the degree of coupling between the individual coordinates. In general, the less coupling the better, as variation of one particular coordinate will then have minimal impact on the other coordinates. Coupling manifests itself primarily as relatively large partial derivative terms between different coordinates. For example, a strong harmonic coupling between two different coordinates,  and
and 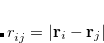 , results in a large off-diagonal element,
, results in a large off-diagonal element,  , in the Hessian (second derivative) matrix. Normally this is the only type of coupling that can be directly “observed” during an optimization, as third and higher derivatives are ignored in almost all optimization algorithms.
, in the Hessian (second derivative) matrix. Normally this is the only type of coupling that can be directly “observed” during an optimization, as third and higher derivatives are ignored in almost all optimization algorithms.
In the early days of computational quantum chemistry geometry optimizations were carried out in Cartesian coordinates. Cartesians are an obvious choice as they can be defined for all systems and gradients and second derivatives are calculated directly in Cartesian coordinates. Unfortunately, Cartesians normally make a poor coordinate set for optimization as they are heavily coupled. Cartesians have been returning to favor later on because of their very general nature, and because it has been clearly demonstrated that if reliable second derivative information is available (i.e., a good starting Hessian) and the initial geometry is reasonable, then Cartesians can be as efficient as any other coordinate set for small to medium-sized molecules.[Baker and Hehre(1991), Baker and Bergeron(1993)] Without good Hessian data, however, Cartesians are inefficient, especially for long chain acyclic systems.
In the 1970s Cartesians were replaced by Z-matrix coordinates. Initially the Z-matrix was used simply as a means of geometry input; it is far easier to describe a molecule in terms of bond lengths, bond angles and dihedral angles (the natural way a chemist thinks of molecular structure) than to develop a suitable set of Cartesian coordinates. It was subsequently found that optimization was generally more efficient in Z-matrix coordinates than in Cartesians, especially for acyclic systems. This is not always the case, and care must be taken in constructing a suitable Z-matrix. A good general rule is ensure that each variable is defined in such a way that changing its value will not change the values of any of the other variables. A brief discussion concerning good Z-matrix construction strategy is given by Schlegel.[Schlegel(1984)]
In 1979 Pulay et al. published a key paper, introducing what were termed natural internal coordinates into geometry optimization.[Pulay et al.(1979)Pulay, Fogarasi, Pang, and Boggs] These coordinates involve the use of individual bond displacements as stretching coordinates, but linear combinations of bond angles and torsions as deformational coordinates. Suitable linear combinations of bends and torsions (the two are considered separately) are selected using group theoretical arguments based on local pseudo-symmetry. For example, bond angles around an  hybridized carbon atom are all approximately tetrahedral, regardless of the groups attached, and idealized tetrahedral symmetry can be used to generate deformational coordinates around the central carbon atom.
hybridized carbon atom are all approximately tetrahedral, regardless of the groups attached, and idealized tetrahedral symmetry can be used to generate deformational coordinates around the central carbon atom.
The major advantage of natural internal coordinates in geometry optimization is their ability to significantly reduce the coupling, both harmonic and anharmonic, between the various coordinates. Compared to natural internals, Z-matrix coordinates arbitrarily omit some angles and torsions (to prevent redundancy), and this can induce strong anharmonic coupling between the coordinates, especially with a poorly constructed Z-matrix. Another advantage of the reduced coupling is that successful minimizations can be carried out in natural internals with only an approximate (e.g., diagonal) Hessian provided at the starting geometry. A good starting Hessian is still needed for a transition state search.
Despite their clear advantages, natural internals have only become used widely at a later stage. This is because, when used in the early programs, it was necessary for the user to define them. This situation changed in 1992 with the development of computational algorithms capable of automatically generating natural internals from input Cartesians.[Fogarasi et al.(1992)Fogarasi, Zhou, Taylor, and Pulay] For minimization, natural internals have become the coordinates of first choice.[Fogarasi et al.(1992)Fogarasi, Zhou, Taylor, and Pulay, Baker and Bergeron(1993)]
There are some disadvantages to natural internal coordinates as they are commonly constructed and used:
Algorithms for the automatic construction of natural internals are complicated. There are a large number of structural possibilities, and to adequately handle even the most common of them can take several thousand lines of code.
For the more complex molecular topologies, most assigning algorithms generate more natural internal coordinates than are required to characterize all possible motions of the system (i.e., the generated coordinate set contains redundancies).
In cases with a very complex molecular topology (e.g., multiply fused rings and cage compounds) the assigning algorithm may be unable to generate a suitable set of coordinates.
The redundancy problem has been addressed in an excellent paper by Pulay and Fogarasi,[Pulay and Fogarasi(1992)] who have developed a scheme for carrying out geometry optimization directly in the redundant coordinate space.
Baker et al.[Baker et al.(1996)Baker, Kessi, and Delley] developed a set of delocalized internal coordinates that eliminate all of the above-mentioned difficulties. Building on some of the ideas in the redundant optimization scheme of Pulay and Fogarasi,[Pulay and Fogarasi(1992)] delocalized internals form a complete, non-redundant set of coordinates which are as good as, if not superior to, natural internals, and which can be generated in a simple and straightforward manner for essentially any molecular topology, no matter how complex.
Consider a set of  internal coordinates
internal coordinates  Displacements
Displacements  in
in  are related to the corresponding Cartesian displacements
are related to the corresponding Cartesian displacements  by means of the usual
by means of the usual 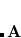 -matrix,[Wilson et al.(1955)Wilson, Decius, and Cross]
-matrix,[Wilson et al.(1955)Wilson, Decius, and Cross]
 |
(A.15) |
If any of the internal coordinates are redundant, then the rows of the -matrix will be linearly dependent.
Delocalized internal coordinates are obtained simply by constructing and diagonalizing the matrix  . Diagonalization of
. Diagonalization of  results in two sets of eigenvectors; a set of
results in two sets of eigenvectors; a set of 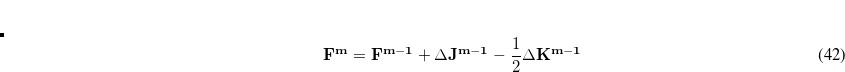 (typically
(typically  , where
, where  is the number of atoms) eigenvectors with eigenvalues
is the number of atoms) eigenvectors with eigenvalues  , and a set of
, and a set of  eigenvectors with eigenvalues
eigenvectors with eigenvalues 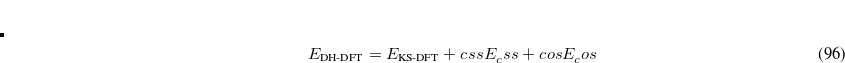 (to numerical precision). In this way, any redundancies present in the original coordinate set are isolated (they correspond to those eigenvectors with zero eigenvalues). The eigenvalue equation of can thus be written
(to numerical precision). In this way, any redundancies present in the original coordinate set are isolated (they correspond to those eigenvectors with zero eigenvalues). The eigenvalue equation of can thus be written
 |
(A.16) |
where  is the set of non-redundant eigenvectors of (those with
is the set of non-redundant eigenvectors of (those with  ) and
) and  is the corresponding redundant set.
is the corresponding redundant set.
The nature of the original set of coordinates is unimportant, as long as it spans all the degrees of freedom of the system under consideration. We include in , all bond stretches, all planar bends and all proper torsions that can be generated based on the atomic connectivity. These individual internal coordinates are termed primitives. This blanket approach generates far more primitives than are necessary, and the set contains much redundancy. This is of little concern, as solution of Eq. (A.16) takes care of all redundancies.
Note that eigenvectors in both and will each be linear combinations of potentially all the original primitives. Despite this apparent complexity, we take the set of non-redundant vectors as our working coordinate set. Internal coordinates so defined are much more delocalized than natural internal coordinates (which are combinations of a relatively small number of bends or torsions) hence, the term delocalized internal coordinates.
It may appear that because delocalized internals are such a complicated mixing of the original primitive internals, they are a poor choice for use in an actual optimization. On the contrary, arguments can be made that delocalized internals are, in fact, the “best” possible choice, certainly at the starting geometry. The interested reader is referred to the original literature for more details.[Baker et al.(1996)Baker, Kessi, and Delley]
The situation for geometry optimization, comparing Cartesian, Z-matrix and delocalized internal coordinates, and assuming a “reasonable” starting geometry, is as follows:
For small or very rigid medium-sized systems (up to about 15 atoms), optimizations in Cartesian and internal coordinates (“good” Z-matrix or delocalized internals) should perform similarly.
For medium-sized systems (say 15–30 atoms) optimizations in Cartesians should perform as well as optimizations in internal coordinates, provided a reliable starting Hessian is available.
For large systems (30+ atoms), unless these are very rigid, neither Cartesian nor Z-matrix coordinates can compete with delocalized internals, even with good quality Hessian information. As the system increases, and with less reliable starting geometries, the advantage of delocalized internals can only increase.
There is one particular situation in which Cartesian coordinates may be the best choice. Natural internal coordinates (and by extension delocalized internals) show a tendency to converge to low energy structures.[Baker and Bergeron(1993)] This is because steps taken in internal coordinate space tend to be much larger when translated into Cartesian space, and, as a result, higher energy local minima tend to be “jumped over”, especially if there is no reliable Hessian information available (which is generally not needed for a successful optimization). Consequently, if the user is looking for a local minimum (i.e., a meta-stable structure) and has both a good starting geometry and a decent Hessian, the user should carry out the optimization in Cartesian coordinates.