A.3 Eigenvector-Following (EF) Algorithm
The work of Cerjan and Miller,[Cerjan and Miller(1981)] and later Simons and co-workers,[Simons et al.(1983)Simons, Jørgensen, Taylor, and Ozment, Banerjee et al.(1985)Banerjee, Adams, Simons, and Shepard] showed that there was a better step than simply directly following one of the Hessian eigenvectors. A simple modification to the Newton-Raphson step is capable of guiding the search away from the current region towards a stationary point with the required characteristics. This is
 |
(A.6) |
in which 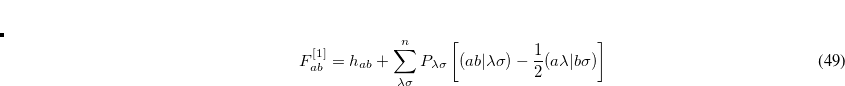 can be regarded as a shift parameter on the Hessian eigenvalue
can be regarded as a shift parameter on the Hessian eigenvalue  . Scaling the Newton-Raphson step in this manner effectively directs the step to lie primarily, but not exclusively (unlike Poppinger’s algorithm[Poppinger(1975)]), along one of the local eigenmodes, depending on the value chosen for . References Cerjan:1981,Simons:1983,Banerjee:1985 all use the same basic approach of Eq. (A.6) but differ in the means of determining the value of .
. Scaling the Newton-Raphson step in this manner effectively directs the step to lie primarily, but not exclusively (unlike Poppinger’s algorithm[Poppinger(1975)]), along one of the local eigenmodes, depending on the value chosen for . References Cerjan:1981,Simons:1983,Banerjee:1985 all use the same basic approach of Eq. (A.6) but differ in the means of determining the value of .
The EF algorithm[Baker(1986)] uses the rational function approach presented in Refs. Banerjee:1985, yielding an eigenvalue equation of the form
 |
(A.7) |
from which a suitable can be obtained. Expanding Eq. (A.7) yields
 |
(A.8) |
and
 |
(A.9) |
In terms of a diagonal Hessian representation, Eq. (A.8) rearranges to Eq. (A.6), and substitution of Eq. (A.6) into the diagonal form of Eq. (A.9) gives
 |
(A.10) |
which can be used to evaluate iteratively.
The eigenvalues, , of the RFO equation Eq. (A.7) have the following important properties:[Banerjee et al.(1985)Banerjee, Adams, Simons, and Shepard]
The
 values of bracket the
values of bracket the  eigenvalues of the Hessian matrix
eigenvalues of the Hessian matrix  .
. At a stationary point, one of the eigenvalues,
, of Eq. (A.7) is zero and the other eigenvalues are those of the Hessian at the stationary point. For a saddle point of order
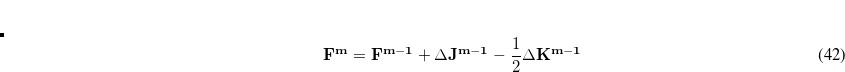 , the zero eigenvalue separates the negative and the
, the zero eigenvalue separates the negative and the  positive Hessian eigenvalues.
positive Hessian eigenvalues.
This last property, the separability of the positive and negative Hessian eigenvalues, enables two shift parameters to be used, one for modes along which the energy is to be maximized and the other for which it is minimized. For a transition state (a first-order saddle point), in terms of the Hessian eigenmodes, we have the two matrix equations
 |
(A.11) |
 |
(A.12) |
where it is assumed that we are maximizing along the lowest Hessian mode  . Note that
. Note that  is the highest eigenvalue of Eq. (A.11), which is always positive and approaches zero at convergence, and
is the highest eigenvalue of Eq. (A.11), which is always positive and approaches zero at convergence, and  is the lowest eigenvalue of Eq. (A.12), which it is always negative and again approaches zero at convergence.
is the lowest eigenvalue of Eq. (A.12), which it is always negative and again approaches zero at convergence.
Choosing these values of gives a step that attempts to maximize along the lowest Hessian mode, while at the same time minimizing along all the other modes. It does this regardless of the Hessian eigenvalue structure (unlike the Newton-Raphson step). The two shift parameters are then used in Eq. (A.6) to give the final step
 |
(A.13) |
If this step is greater than the maximum allowed, it is scaled down. For minimization only one shift parameter, , would be used which would act on all modes.
In Eq. (A.11) and Eq. (A.12) it was assumed that the step would maximize along the lowest Hessian mode, 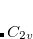 , and minimize along all the higher modes. However, it is possible to maximize along modes other than the lowest, and in this way perhaps locate transition states for alternative rearrangements/dissociations from the same initial starting point. For maximization along the
, and minimize along all the higher modes. However, it is possible to maximize along modes other than the lowest, and in this way perhaps locate transition states for alternative rearrangements/dissociations from the same initial starting point. For maximization along the 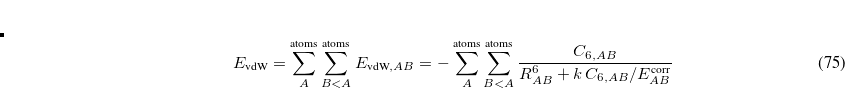 th mode (instead of the lowest mode), Eq. (A.11) is replaced by
th mode (instead of the lowest mode), Eq. (A.11) is replaced by
 |
(A.14) |
and Eq. (A.12) would now exclude the th mode but include the lowest. Since what was originally the th mode is the mode along which the negative eigenvalue is required, then this mode will eventually become the lowest mode at some stage of the optimization. To ensure that the original mode is being followed smoothly from one cycle to the next, the mode that is actually followed is the one with the greatest overlap with the mode followed on the previous cycle. This procedure is known as mode following. For more details and some examples, see Ref. Baker:1986.