3.5 Additional Input Sections
The $molecule and $rem sections are required for all Q-Chem jobs, but depending on the details of the job a number of other input sections may be required. These are summarized briefly below, with references to more detailed descriptions to be found later in this manual.
3.5.1 Comments ($comment)
Users are able to add comments to the input file outside keyword input sections, which will be ignored by the program. This can be useful as reminders to the user, or perhaps, when teaching another user to set up inputs. Comments can also be provided in a $comment block, which is actually redundant given that the entire input deck is copied to the output file.
3.5.2 User-Defined Basis Sets ($basis and $aux_basis)
By setting the $rem keyword BASIS = GEN, the user indicates that the basis set will be user defined. In that case, the $basis input section is used to specify the basis set. Similarly, if AUX_BASIS = GEN then the $aux_basis input section is used to specify the auxiliary basis set. See Chapter 8 for details on how to input a user-defined basis set.
3.5.3 User-Defined Effective Core Potential ($ecp)
By setting ECP = GEN, the user indicates that the effective core potentials (pseudopotentials, which replace explicit core electrons) to be used will be defined by the user. In that case, the $ecp section is used to specify these pseudopotentials. See Chapter 9 for further details.
3.5.4 User-Defined Exchange-Correlation Density Functionals
($xc_functional)
If the keyword EXCHANGE = GEN then a DFT calculation will be performed using a user-specified combination of exchange and correlation functional(s), as described in Chapter 4. Custom functionals of this sort can be constructed as any linear combination of exchange and/or correlation functionals that are supported by Q-Chem; see Section 5.4 for a list of supported functionals. The format for the $xc_functional input section is the following:
$xc_functional
X exchange_symbol coefficient
X exchange_symbol coefficient
...
C correlation_symbol coefficient
C correlation_symbol coefficient
...
K coefficient
$end
Note: The coefficients must be real numbers.
3.5.5 User-defined Parameters for DFT Dispersion Correction
($empirical_dispersion)
If a user wants to change from the default values recommended by Grimme, then user-defined parameters can be specified using the $empirical_dispersion input section. See Section 5.7.2 for details.
3.5.6 Addition of External Point Charges ($external_charges)
If the $external_charges keyword is present, Q-Chem scans for a set of external charges to be incorporated into a calculation. The format is shown below and consists of Cartesian coordinates and the value of the point charge, with one charge per line. The charge is in atomic units and the coordinates are in Ångstroms, unless bohrs are selected by setting the $rem keyword INPUT_BOHR to TRUE. The external charges are rotated with the molecule into the standard nuclear orientation.
Example 3.14 General format for incorporating a set of external charges.
$external_charges
x-coord1 y-coord1 z-coord1 charge1
x-coord2 y-coord2 z-coord2 charge2
x-coord3 y-coord3 z-coord3 charge3
$end
In addition, the user can request to add a charged cage around the molecule (for so-called “charge stabilization” calculations) using the keyword ADD_CHARGED_CAGE. See Section 7.7.7 for details.
3.5.7 Applying a Multipole Field ($multipole_field)
A multipole field can be applied to the molecule under investigation by specifying the $multipole_field input section. Each line in this section consists of a single component of the applied field, in the following format.
Example 3.15 General format for imposing a multipole field.
$multipole_field
field_component_1 value_1
field_component_2 value_2
$end
Each field_component is stipulated using the Cartesian representation e.g., X, Y, and/or Z, (dipole field components); XX, XY, and/or YY (quadrupole field components); XXX, XXY, etc.. The value (magnitude) of each field component should be provided in atomic units.
3.5.8 User-Defined Occupied Guess Orbitals ($occupied and
$swap_occupied_virtual)
It is sometimes useful for the occupied guess orbitals to be different from the lowest 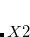 (or
(or 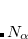 ) orbitals. Q-Chem allows the occupied guess orbitals to be defined using the $occupied keyword. Using the $occupied input section, the user can choose which orbitals (by number) to occupy by specifying the
) orbitals. Q-Chem allows the occupied guess orbitals to be defined using the $occupied keyword. Using the $occupied input section, the user can choose which orbitals (by number) to occupy by specifying the 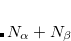 -spin orbitals on the first line of the $occupied section and the
-spin orbitals on the first line of the $occupied section and the  -spin orbitals on the second line. For large molecules where only a few occupied
-spin orbitals on the second line. For large molecules where only a few occupied  virtual promotions are desired, it is simpler to use the $swap_occupied_virtual input section. Details can be found in Section 4.4.4.
virtual promotions are desired, it is simpler to use the $swap_occupied_virtual input section. Details can be found in Section 4.4.4.
3.5.9 Polarizable Continuum Solvation Models ($pcm)
The $pcm section provides fine-tuning of the job control for polarizable continuum models (PCMs), which are requested by setting the $rem keyword SOLVENT_METHOD equal to PCM. Supported PCMs include C-PCM, IEF-PCM, and SS(V)PE, which share a common set of job-control variables. Details are provided in Section 12.2.2.
3.5.10 SS(V)PE Solvation Modeling ($svp and $svpirf)
The $svp section is available to specify special parameters to the solvation module such as cavity grid parameters and modifications to the numerical integration procedure. The $svpirf section allows the user to specify an initial guess for the solution of the cavity charges. As discussed in section 12.2.5, the $svp and $svpirf input sections are used to specify parameters for the iso-density implementation of SS(V)PE. An alternative implementation of the SS(V)PE mode, based on a more empirical definition of the solute cavity, is available in the PCM (see Section 12.2.2) and controlled from within the $pcm input section.
3.5.11 User-Defined van der Waals Radii ($van_der_waals)
The $van_der_waals section of the input enables the user to customize the van der Waals radii that are important parameters in the Langevin dipoles solvation model; see Section 12.2.
3.5.12 Effective Fragment Potential Calculations ($efp_fragments and $efp_params)
These keywords are used to specify positions and parameters for effective fragments in EFP calculations. Details are provided in Section 12.5.
3.5.13 Natural Bond Orbital Package ($nbo)
When NBO is set to TRUE in the $rem section, a natural bond orbital (NBO) calculation is performed, using the Q-Chem interface to the NBO 5.0 and NBO 6.0 packages. In such cases, the $nbo section may contain standard parameters and keywords for the NBO program.
3.5.14 Orbitals, Densities and Electrostatic Potentials on a Mesh ($plots)
The $plots part of the input permits the evaluation of molecular orbitals, densities, electrostatic potentials, transition densities, electron attachment and detachment densities on a user-defined mesh of points. Q-Chem will print out the raw data, but can also format these data into the form of a “cube” file that is a standard input format for volumetric data that can be read various visualization programs. See Section 11.5 for details.
3.5.15 Intracules ($intracule)
Setting the $rem keyword INTRACULE = TRUE requests a molecular intracule calculation, in which case additional customization is possible using the $intracule input section. See Section 11.9.
3.5.16 Geometry Optimization with Constraints ($opt)
For JOBTYPE = OPT, Q-Chem scans the input file for the $opt section. Here, the user may specify distance, angle, dihedral and out-of-plane bend constraints to be imposed on the optimization procedure, as described in Chapter 10.
3.5.17 Isotopic Substitutions ($isotopes)
For vibrational frequency calculations (JOBTYPE = FREQ), nuclear masses are set by default to be those corresponding to the most abundant naturally-occurring isotopes. Alternative masses for one or more nuclei can be requested by setting ISOTOPES = TRUE in the $rem section, in which case the $isotopes section is used to specify the desired masses as described in Section 11.10.2. Isotopic substitutions incur negligible additional cost in a frequency calculation.