7.7 Auxiliary Basis Sets for RI (Density Fitting)
While atomic orbital standard basis sets are used to expand one-electron functions such as molecular orbitals, auxiliary basis sets are also used in many Q-Chem jobs to efficiently approximate products of one-electron functions, such as arise in electron correlation methods.
For a molecule of fixed size, increasing the number of basis functions per atom, 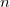 , leads to
, leads to 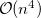 growth in the number of significant four-center two-electron integrals, since the number of non-negligible product charge distributions,
growth in the number of significant four-center two-electron integrals, since the number of non-negligible product charge distributions, 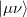 , grows as
, grows as 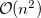 . As a result, the use of large (high-quality) basis expansions is computationally costly. Perhaps the most practical way around this “basis set quality” bottleneck is the use of auxiliary basis expansions [324, 325, 326]. The ability to use auxiliary basis sets to accelerate a variety of electron correlation methods, including both energies and analytical gradients, is a major feature of Q-Chem.
. As a result, the use of large (high-quality) basis expansions is computationally costly. Perhaps the most practical way around this “basis set quality” bottleneck is the use of auxiliary basis expansions [324, 325, 326]. The ability to use auxiliary basis sets to accelerate a variety of electron correlation methods, including both energies and analytical gradients, is a major feature of Q-Chem.
The auxiliary basis 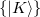 is used to approximate products of Gaussian basis functions:
is used to approximate products of Gaussian basis functions:
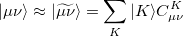 |
(7.1) |
Auxiliary basis expansions were introduced long ago, and are now widely recognized as an effective and powerful approach, which is sometimes synonymously called resolution of the identity (RI) or density fitting (DF). When using auxiliary basis expansions, the rate of growth of computational cost of large-scale electronic structure calculations with is reduced to approximately 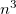 .
.
If is fixed and molecule size increases, auxiliary basis expansions reduce the pre-factor associated with the computation, while not altering the scaling. The important point is that the pre-factor can be reduced by 5 or 10 times or more. Such large speedups are possible because the number of auxiliary functions required to obtain reasonable accuracy, 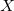 , has been shown to be only about 3 or 4 times larger than
, has been shown to be only about 3 or 4 times larger than 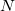 .
.
The auxiliary basis expansion coefficients,  , are determined by minimizing the deviation between the fitted distribution and the actual distribution,
, are determined by minimizing the deviation between the fitted distribution and the actual distribution, 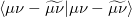 , which leads to the following set of linear equations:
, which leads to the following set of linear equations:
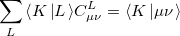 |
(7.2) |
Evidently solution of the fit equations requires only two- and three-center integrals, and as a result the (four-center) two-electron integrals can be approximated as the following optimal expression for a given choice of auxiliary basis set:
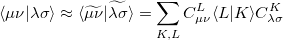 |
(7.3) |
In the limit where the auxiliary basis is complete (i.e. all products of AOs are included), the fitting procedure described above will be exact. However, the auxiliary basis is invariably incomplete (as mentioned above, 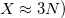 because this is essential for obtaining increased computational efficiency.
because this is essential for obtaining increased computational efficiency.
More details on Q-Chem’s use of RI methods is given in Section 5.6 on RI-MP2 and related methods, Section 5.15 on pairing methods, Section 5.8.5 on coupled cluster methods, Section 4.8.8 on DFT methods, and Section 6.9 on restricted active space methods. In the remainder of this section we focus on documenting the input associated with the auxiliary basis itself.
Q-Chem contains a variety of built-in auxiliary basis sets, that can be specified by the $rem keyword AUX_BASIS.
AUX_BASIS
Sets the auxiliary basis set to be used
TYPE:
STRING
DEFAULT:
No default auxiliary basis set
OPTIONS:
General, Gen
User-defined. As for BASIS
Symbol
Use standard auxiliary basis sets as in the table below
Mixed
Use a combination of different basis sets
RECOMMENDATION:
Consult literature and EMSL Basis Set Exchange to aid your selection.
Symbolic Name |
Atoms Supported |
RIMP2-VDZ |
H, He, Li |
RIMP2-TZVPP |
H, He, Li |
RIMP2-cc-pVDZ |
H, He, Li |
RIMP2-cc-pVTZ |
H, He, Li |
RIMP2-cc-pVQZ |
H, He, Li |
RIMP2-aug-cc-pVDZ |
H, He, B |
RIMP2-aug-cc-pVTZ |
H, He, B |
RIMP2-aug-cc-pVQZ |
H, He, B |
 Ne, Na
Ne, Na In addition to built-in auxiliary basis sets, it is also possible to enter user-defined auxiliary basis sets using an $aux_basis input section, whose syntax generally follows the $basis input section documented above in Section 7.4.