6.9 Restricted Active Space Spin-Flip (RAS-SF) and Configuration Interaction (RAS-CI)
The restricted active space spin-flip (RAS-SF) is a special form of configuration interaction that is capable of describing the ground and low-lying excited states with moderate computational cost in a single-reference formulation [500, 501, 502, 503], including strongly correlated systems. The RAS-SF approach is essentially a much lower computational cost alternative to Complete Active Space SCF (CAS-SCF) methods. RAS-SF typically works by performing a full CI calculation within an active space that is defined by the half-occupied orbitals of a restricted open shell HF (ROHF) reference determinant. In this way the difficulties of state-specific orbital optimization in CAS-SCF are bypassed. Single excitations into (hole) and out of (particle) the active space provide state-specific relaxation instead. Unlike most CI-based methods, RAS-SF is size-consistent, as well as variational, and, the increase in computational cost with system size is modest for a fixed number of spin flips. Beware, however, for the increase in cost as a function of the number of spin-flips is exponential! RAS-SF has been shown to be capable of tackling multiple low-lying electronic states in polyradicals and reliably predicting ground state multiplicities [500, 504, 505, 506, 501, 502].
RAS-SF can also be viewed as one particular case of a more general RAS-CI family of methods. For instance, instead of defining the active space via spin-flipping as above, initial orbitals of other types can be read in, and electronic excitations calculated this way may be viewed as a RAS-EE-CI method (though size-consistency will generally be lost). Similar to EOM-CC approaches (see Section 6.7), other target RAS-CI wave functions can be constructed starting from any electronic configuration as the reference and using a general excitation-type operator. For instance, one can construct an ionizing variant that removes an arbitrary number of particles that is RAS-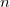 IP-CI. An electron attaching variant is RAS-EA-CI [503].
IP-CI. An electron attaching variant is RAS-EA-CI [503].
Q-Chem features two versions of RAS-CI code with different, complementary, functionality. One code (invoked by specifying CORRRELATION = RASCI) has been written by David Casanova [503]; below we will refer to this code as RASCI1. The second implementation (invoked by specifying CORRRELATION = RASCI2) is primarily by Paul Zimmerman [501]; we will refer to it as RASCI2 below.
The RASCI1 code uses an integral-driven implementation (exact integrals) and spin-adaptation of the CI configurations which results in a smaller diagonalization dimension. The current Q-Chem implementation of RASCI1 only allows for the calculation of systems with an even number of electrons, with the multiplicity of each state being printed alongside the state energy. Shared memory parallel execution decreases compute time as all the underlying integrals routines are parallelized.
The RASCI2 code includes the ability to simulate closed and open shell systems (i.e., even and odd numbers of electrons), fast integral evaluation using the resolution of the identity (RI) approximation, shared memory parallel operation, and analysis of the 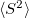 values and natural orbitals. The natural orbitals are stored in the QCSCRATCH directory in a folder called “NOs” in MolDen-readable format. Shared memory parallel is invoked as described in Section 2.8. A RASCI2 input requires the specification of an auxiliary basis set analogous to RI-MP2 computations (see Section 5.6.1). Otherwise, the active space as well as hole and particle excitations are specified in the same way as in RASCI1.
values and natural orbitals. The natural orbitals are stored in the QCSCRATCH directory in a folder called “NOs” in MolDen-readable format. Shared memory parallel is invoked as described in Section 2.8. A RASCI2 input requires the specification of an auxiliary basis set analogous to RI-MP2 computations (see Section 5.6.1). Otherwise, the active space as well as hole and particle excitations are specified in the same way as in RASCI1.
Note: Because RASCI2 uses the RI approximation, the total energies computed with the two codes will be slightly different; however, the energy differences between different states should closely match each other.
6.9.1 The Restricted Active Space (RAS) Scheme
In the RAS formalism, we divide the orbital space into three subspaces called RAS1, RAS2 and RAS3 (Fig. 6.2). The RAS-CI states are defined by the number of orbitals and the restrictions in each subspace.

The single reference RAS-CI electronic wave functions are obtained by applying a spin-flipping or excitation operator 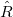 on the reference determinant
on the reference determinant 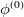 .
.
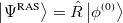 |
(6.83) |
The operator must obey the restrictions imposed in the subspaces RAS1, RAS2 and RAS3, and can be decomposed as:
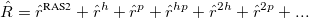 |
(6.84) |
where 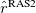 contains all possible electronic promotions within the RAS2 space, that is, a reduced full CI, and the rest of the terms generate configurations with different number of holes (
contains all possible electronic promotions within the RAS2 space, that is, a reduced full CI, and the rest of the terms generate configurations with different number of holes (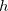 super-index) in RAS1 and electrons in RAS3 (
super-index) in RAS1 and electrons in RAS3 (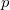 super-index). The current implementation truncates this series up to the inclusion of hole and particle contributions, i.e. the first three terms on the right hand side of Eq. (6.84).
super-index). The current implementation truncates this series up to the inclusion of hole and particle contributions, i.e. the first three terms on the right hand side of Eq. (6.84).
6.9.2 Second-Order Perturbative Corrections to RAS-CI
In general, the RAS-CI family of methods within the 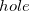 and
and 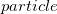 approximation is unable to capture the necessary amounts of dynamic correlation for the computation of relative energies with chemical accuracy. The missed correlation can be added on top of the RAS-CI wave function using multi-reference perturbation theory (MRPT).[507] The second order energy correction, i.e. RASCI(2), can be expressed as:
approximation is unable to capture the necessary amounts of dynamic correlation for the computation of relative energies with chemical accuracy. The missed correlation can be added on top of the RAS-CI wave function using multi-reference perturbation theory (MRPT).[507] The second order energy correction, i.e. RASCI(2), can be expressed as:
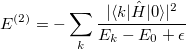 |
(6.85) |
where 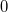 indicates the zero-order space and
indicates the zero-order space and 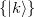 is the complementary set of determinants. There is no natural choice for the
is the complementary set of determinants. There is no natural choice for the 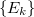 excited energies in MRPT, and two different models are available within the RASCI(2) approach, that is the Davidson-Kapuy and Epstein-Nesbet partitionings. As it is common practice in many second-order MRPT corrections, the denominator energy differences in Eq. (6.85) can be level shifted with a parameter
excited energies in MRPT, and two different models are available within the RASCI(2) approach, that is the Davidson-Kapuy and Epstein-Nesbet partitionings. As it is common practice in many second-order MRPT corrections, the denominator energy differences in Eq. (6.85) can be level shifted with a parameter  .
.
6.9.3 Excitonic Analysis of the RAS-CI Wave Function
The RAS-CI wave function of multi-chromophoric systems can be decomposed in terms of local excitations (LE), charge resonances (CR) and multi-exciton configurations (ME).
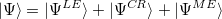 |
(6.86) |
The wave function decomposition scheme combines the use of fragment (localized) orbitals with the computation of charge and spin cumulant indexes.[508, 509] Such analysis is extremely useful in order to characterize in great detail the nature of the ground and excited states in the presence of two or more molecules or chromophoric moieties. The ME contributions can be further decomposed into different spin coupled configurations localized on each. Typically, the ME part of a singlet state in a molecular dimer can be decomposed as singlet-singlet (SS) and triplet-tripled (TT) contributions.
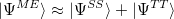 |
(6.87) |
At the moment, this analysis is only available for the decomposition of spin singlet states.
6.9.4 Job Control for the RASCI1 Implementation
At present the RASCI1 and RASCI2 implementations employ different keywords (which will be reconciled in a future version). This subsection applies to RASCI1 (even electron systems, spin adapted algorithm using exact integrals).
The use of the RAS-CI1 methodology is controlled by setting the $rem variable CORRELATION to RASCI and EXCHANGE should be set to HF. The RASCI1 implementation is only compatible with even numbers of electrons and restricted orbitals, i.e., UNRESTRICTED = FALSE.
The minimum input also requires specifying the desired (non-zero) value for RAS_ROOTS, the number of electrons in the “active” RAS2 space and the number of orbitals in RAS1 and RAS2 subspaces.
RAS_ROOTS
Sets the number of RAS-CI roots to be computed.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
Compute
RECOMMENDATION:
None. Only works with RASCI.
RAS_ELEC
Sets the number of electrons in RAS2 (active electrons).
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
User-defined integer,
RECOMMENDATION:
None. Only works with RASCI.
RAS_ACT
Sets the number of orbitals in RAS2 (active orbitals).
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
User-defined integer,
RECOMMENDATION:
None. Only works with RASCI.
RAS_OCC
Sets the number of orbitals in RAS1
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined integer,
RECOMMENDATION:
These are the initial doubly occupied orbitals (RAS1) before including
RAS_DO_HOLE
Controls the presence of hole excitations in the RAS-CI wave function.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Include hole configurations (RAS1 to RAS2 excitations)
FALSE
Do not include hole configurations
RECOMMENDATION:
None. Only works with RASCI.
RAS_DO_PART
Controls the presence of particle excitations in the RAS-CI wave function.
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
Include particle configurations (RAS2 to RAS3 excitations)
FALSE
Do not include particle configurations
RECOMMENDATION:
None. Only works with RASCI.
RAS_AMPL_PRINT
Defines the absolute threshold (
) for the CI amplitudes to be printed.
TYPE:
INTEGER
DEFAULT:
10
0.1 minimum absolute amplitude
OPTIONS:
User-defined integer,
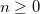
RECOMMENDATION:
None. Only works with RASCI.
RAS_ACT_ORB
Sets the user-selected active orbitals (RAS2 orbitals).
TYPE:
INTEGER ARRAY
DEFAULT:
From RAS_OCC+1 to RAS_OCC+RAS_ACT
OPTIONS:
![$[i,j,k...]$](images/img-1087.png)
The number of orbitals must be equal to the RAS_ACT variable
RECOMMENDATION:
None. Only works with RASCI.
RAS_NATORB
Controls the computation of the natural orbital occupancies.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Compute natural orbital occupancies for all states
FALSE
Do not compute natural orbital occupancies
RECOMMENDATION:
None. Only works with RASCI.
RAS_NATORB_STATE
Allows to save the natural orbitals of a RAS-CI computed state.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:

Saves the natural orbitals for the
RECOMMENDATION:
None. Only works with RASCI.
RAS_GUESS_CS
Controls the number of closed shell guess configurations in RAS-CI.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Imposes to start with
RECOMMENDATION:
Only relevant for the computation of singlet states. Only works with RASCI.
RAS_SPIN_MULT
Specifies the spin multiplicity of the roots to be computed
TYPE:
INTEGER
DEFAULT:
1
Singlet states
OPTIONS:
0
Compute any spin multiplicity
User-defined integer,
RECOMMENDATION:
Only for RASCI, which at present only allows for the computation of systems with an even number of electrons. Thus, RAS_SPIN_MULT only can take odd values.
RAS_PT2
Perform second-order perturbative correction to RAS-CI energy
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
Compute RASCI(2) energy corrections
FALSE
Do not compute RASCI(2) energy corrections
RECOMMENDATION:
None. Only works with RASCI.
RAS_PT2_PARTITION
Specifies the partitioning scheme in RASCI(2)
TYPE:
INTEGER
DEFAULT:
1
Davidson-Kapuy (DK) partitioning
OPTIONS:
2
Epstein-Nesbet (EN) partitioning
0
Do both DK and EN partitionings
RECOMMENDATION:
Only for RASCI if RAS_PT2 is set to true.
RAS_PT2_VSHIFT
Defines the energy level shift (
) in RASCI(2)
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
User-defined integer
RECOMMENDATION:
Only for RASCI if RAS_PT2 is set to true.
RAS_NFRAG
If
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Number of fragments to be considered
RECOMMENDATION:
Only for RASCI. The printed information level is controlled by RAS_PRINT.
RAS_NFRAG_ATOMS
Sets the number of atoms in each fragment.
TYPE:
INTEGER ARRAY
DEFAULT:
None
OPTIONS:
The sum of the numbers must be equal to the total number of atoms in the systems
RECOMMENDATION:
None. Only works with RASCI.
RAS_FRAG_MO
Sets the number of atoms in each fragment.
TYPE:
INTEGER ARRAY
DEFAULT:
[NOcc,NAct,NVir]
Number of orbitals within RAS1, RAS2 and RAS3 spaces
OPTIONS:
Defines sets of canonical MOs to be localized into
RECOMMENDATION:
Setting within RAS1, RAS2 and RAS3 spaces alleviates the computational cost of the localization procedure. It might also result in improved fragment orbitals. Only works with RASCI.
6.9.5 Job Control Options for RASCI2
At present the RASCI1 and RASCI2 implementations employ different keywords (which will be reconciled in a future version). This subsection applies to RASCI2 (even and odd electron systems, determinant-driven algorithm using the resolution of the identity approximation).
The use of the RAS-CI2 methodology is controlled by setting the CORRELATION = RASCI2 and EXCHANGE = HF. The minimum input also requires specifying the desired (non-zero) value for RAS_N_ROOTS, and the number of active occupied and virtual orbital comprising the “active” RAS2 space. RASCI2 calculations also require specification of an auxiliary basis via AUX_BASIS.
RAS_N_ROOTS
Sets the number of RAS-CI roots to be computed.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
RECOMMENDATION:
None. Only works with RASCI2
RAS_ACT_OCC
Sets the number of occupied orbitals to enter the RAS active space.
TYPE:
Integer
DEFAULT:
None
OPTIONS:
n
user defined integer
RECOMMENDATION:
None. Only works with RASCI2
RAS_ACT_VIR
Sets the number of virtual orbitals to enter the RAS active space.
TYPE:
Integer
DEFAULT:
None
OPTIONS:
n
user defined integer
RECOMMENDATION:
None. Only works with RASCI2.
RAS_ACT_DIFF
Sets the number of alpha vs. beta electrons
TYPE:
Integer
DEFAULT:
None
OPTIONS:
n
user defined integer
RECOMMENDATION:
Set to 1 for an odd number of electrons or a cation, -1 for an anion. Only works with RASCI2.
Other $rem variables that can be used to control the evaluation of RASCI2 calculations are SET_ITER for the maximum number of Davidson iterations, and N_FROZEN_CORE and N_FROZEN_VIRTUAL to exclude core and/or virtual orbitals from the RASCI2 calculation.
6.9.6 Examples
Example 6.186 Input for a RAS-2SF-CI calculation of three states of the DDMX tetraradical using RASCI1. The active space (RAS2) contains 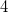 electrons in the singly occupied orbitals in the ROHF quintet reference. Natural orbital occupancies are requested.
electrons in the singly occupied orbitals in the ROHF quintet reference. Natural orbital occupancies are requested.
$molecule
0 5
C 0.000000 0.000000 1.092150
C -1.222482 0.000000 0.303960
C -2.390248 0.000000 1.015958
H -2.344570 0.000000 2.095067
H -3.363161 0.000000 0.537932
C -1.215393 0.000000 -1.155471
H -2.150471 0.000000 -1.702536
C 0.000000 0.000000 -1.769131
C 1.215393 0.000000 -1.155471
H 2.150471 0.000000 -1.702536
C 1.222482 0.000000 0.303960
C 2.390248 0.000000 1.015958
H 2.344570 0.000000 2.095067
H 3.363161 0.000000 0.537932
$end
$rem
EXCHANGE hf
CORRELATION rasci
BASIS 6-31g
UNRESTRICTED false
MEM_TOTAL 4000
MEM_STATIC 100
RAS_ROOTS 3
RAS_ACT 4
RAS_ELEC 4
RAS_OCC 25
RAS_SPIN_MULT 0
RAS_NATORB true
$end
Example 6.187 Input for a RAS-2IP-CI calculation of triplet states of F molecule using the dianion closed shell F
molecule using the dianion closed shell F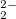 as the reference determinant. RASCI1 code is used
as the reference determinant. RASCI1 code is used
$molecule
-2 1
F
F 1 rFF
rFF = 1.4136
$end
$rem
EXCHANGE hf
CORRELATION rasci
BASIS cc-pVTZ
MEM_TOTAL 4000
MEM_STATIC 100
RAS_ROOTS 2
RAS_ACT 6
RAS_ELEC 10
RAS_OCC 4
RAS_SPIN_MULT 3
$end
Example 6.188 Input for a FCI/6-31G calculation of water molecule expanding the RAS2 space to the entire molecular orbital set. RASCI code is used.
$molecule
0 1
O 0.000 0.000 0.120
H -0.762 0.000 -0.479
H 0.762 0.000 -0.479
$end
$rem
EXCHANGE hf
CORRELATION rasci
BASIS 6-31G
MEM_TOTAL 4000
MEM_STATIC 100
RAS_ROOTS 1
RAS_ACT 13
RAS_ELEC 10
RAS_OCC 0
RAS_SPIN_MULT 1
RAS_DO_HOLE false
RAS_DO_PART false
$end
Example 6.189 Methylene single spin-flip calculation using RASCI2
$rem
EXCHANGE HF
CORRELATION RASCI2
BASIS cc-pVDZ
AUX_BASIS rimp2-cc-pVDZ
UNRESTRICTED false
RAS_ACT_OCC 1 ! # alpha electrons
RAS_ACT_VIR 1 ! # virtuals in active space
RAS_ACT_DIFF 0 ! # set to 1 for odd # of e-s
RAS_N_ROOTS 4
SET_ITER 25 ! number of iterations in RASCI
$end
$molecule
0 3
C 0.0000000 0.0000000 0.0000000
H -0.8611113 0.0000000 0.6986839
H 0.8611113 0.0000000 0.6986839
$end
Example 6.190 Two methylene separated by 10 ; double spin-flip calculation using RASCI2. Note that the values for this case will not be uniquely defined at the triply-degenerate ground state.
$rem
EXCHANGE HF
CORRELATION RASCI2
BASIS cc-pVDZ
AUX_BASIS rimp2-cc-pVDZ
RAS_ACT_OCC 2 ! # alpha electrons
RAS_ACT_VIR 2 ! # virtuals in active space
RAS_ACT_DIFF 0 ! # set to 1 for odd # of e-s
UNRESTRICTED false
RAS_N_ROOTS 8
SET_ITER 25
$end
$molecule
0 5
C 0.0000000 0.0000000 0.0000000
H -0.8611113 0.0000000 0.6986839
H 0.8611113 0.0000000 0.6986839
C 0.0000000 10.0000000 0.0000000
H -0.8611113 10.0000000 0.6986839
H 0.8611113 10.0000000 0.6986839
$end
Example 6.191 RASCI2 calculation of the nitrogen cation using double spin-flip.
$rem
EXCHANGE HF
CORRELATION RASCI2
BASIS 6-31G*
AUX_BASIS rimp2-VDZ
RAS_ACT_OCC 3 ! # alpha electrons
RAS_ACT_VIR 3 ! # virtuals in active space
RAS_ACT_DIFF 1 ! # for odd # e-s, cation
UNRESTRICTED false
N_FROZEN_CORE 2
N_FROZEN_VIRTUAL 2
RAS_N_ROOTS 8
SET_ITER 25
$end
$molecule
1 6
N 0.0000000 0.0000000 0.0000000
N 0.0000000 0.0000000 4.5
$end