5.1 Introduction
DFT[Parr and Yang(1989), Kohn et al.(1996)Kohn, Becke, and Parr, Laird et al.(1996)Laird, Ross, and Ziegler, Ziegler(1991)] has emerged as an accurate, alternative first-principles approach to quantum mechanical molecular investigations. DFT calculations account for the overwhelming majority of all quantum chemistry calculations, not only because of its proven chemical accuracy, but also because of its relatively low computational expense, comparable to Hartree-Fock theory but with treatment of electron correlation that is neglected in a HF calculation. These two features suggest that DFT is likely to remain a leading method in the quantum chemist’s toolkit well into the future. Q-Chem contains fast, efficient and accurate algorithms for all popular density functionals, making calculations on large molecules possible and practical.
DFT is primarily a theory of electronic ground state structures based on the electron density, 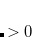 , as opposed to the many-electron wave function,
, as opposed to the many-electron wave function, 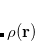 . (Its excited-state extension, time-dependent DFT, is discussed in Section 7.3.) There are a number of distinct similarities and differences between traditional wave function approaches and modern DFT methodologies. First, the essential building blocks of the many-electron wave function
. (Its excited-state extension, time-dependent DFT, is discussed in Section 7.3.) There are a number of distinct similarities and differences between traditional wave function approaches and modern DFT methodologies. First, the essential building blocks of the many-electron wave function 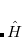 are single-electron orbitals, which are directly analogous to the Kohn-Sham orbitals in the DFT framework. Second, both the electron density and the many-electron wave function tend to be constructed via a SCF approach that requires the construction of matrix elements that are conveniently very similar.
are single-electron orbitals, which are directly analogous to the Kohn-Sham orbitals in the DFT framework. Second, both the electron density and the many-electron wave function tend to be constructed via a SCF approach that requires the construction of matrix elements that are conveniently very similar.
However, traditional ab initio approaches using the many-electron wave function as a foundation must resort to a post-SCF calculation (Chapter 6) to incorporate correlation effects, whereas DFT approaches incorporate correlation at the SCF level. Post-SCF methods, such as perturbation theory or coupled-cluster theory are extremely expensive relative to the SCF procedure. On the other hand, while the DFT approach is exact in principle, in practice it relies on modeling an unknown exchange-correlation energy functional. While more accurate forms of such functionals are constantly being developed, there is no systematic way to improve the functional to achieve an arbitrary level of accuracy. Thus, the traditional approaches offer the possibility of achieving a systematically-improvable level of accuracy, but can be computationally demanding, whereas DFT approaches offer a practical route, but the theory is currently incomplete.