6.9 Non-iterative Corrections to Coupled Cluster Energies
6.9.1 (T) Triples Corrections
To approach chemical accuracy in reaction energies and related properties, it is necessary to account for electron correlation effects that involve three electrons simultaneously, as represented by triple substitutions relative to the mean field single determinant reference, which arise in MP4. The best standard methods for including triple substitutions are the CCSD(T)[Raghavachari et al.(1989)Raghavachari, Trucks, Pople, and Head-Gordon] and QCISD(T) methods.[Pople et al.(1987)Pople, Head-Gordon, and Raghavachari] The accuracy of these methods is well-documented for many cases,[Lee and Scuseria(1995)] and in general is a very significant improvement relative to the starting point (either CCSD or QCISD). The cost of these corrections scales with the 7th power of molecule size (or the 4th power of the number of basis functions, for a fixed molecule size), although no additional disk resources are required relative to the starting coupled-cluster calculation. Q-Chem supports the evaluation of CCSD(T) and QCISD(T) energies, as well as the corresponding OD(T) correction to the optimized doubles method discussed in the previous subsection. Gradients and properties are not yet available for any of these (T) corrections.
6.9.2 (2) Triples and Quadruples Corrections
While the (T) corrections discussed above have been extraordinarily successful, there is nonetheless still room for further improvements in accuracy, for at least some important classes of problems. They contain judiciously chosen terms from 4th- and 5th-order Møller-Plesset perturbation theory, as well as higher order terms that result from the fact that the converged cluster amplitudes are employed to evaluate the 4th- and 5th-order order terms. The (T) correction therefore depends upon the bare reference orbitals and orbital energies, and in this way its effectiveness still depends on the quality of the reference determinant. Since we are correcting a coupled-cluster solution rather than a single determinant, this is an aspect of the (T) corrections that can be improved. Deficiencies of the (T) corrections show up computationally in cases where there are near-degeneracies between orbitals, such as stretched bonds, some transition states, open shell radicals, and diradicals.
Prof. Steve Gwaltney, while working at Berkeley with Martin Head-Gordon, has suggested a new class of non iterative correction that offers the prospect of improved accuracy in problem cases of the types identified above.[Gwaltney and Head-Gordon(2000)] Q-Chem contains Gwaltney’s implementation of this new method, for energies only. The new correction is a true second-order correction to a coupled-cluster starting point, and is therefore denoted as (2). It is available for two of the cluster methods discussed above, as OD(2) and CCSD(2).[Gwaltney and Head-Gordon(2000), Gwaltney and Head-Gordon(2001)] Only energies are available at present.
The basis of the (2) method is to partition not the regular Hamiltonian into perturbed and unperturbed parts, but rather to partition a similarity-transformed Hamiltonian, defined as 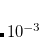 . In the truncated space (call it the
. In the truncated space (call it the  -space) within which the cluster problem is solved (e.g., singles and doubles for CCSD), the coupled-cluster wave function is a true eigenvalue of
-space) within which the cluster problem is solved (e.g., singles and doubles for CCSD), the coupled-cluster wave function is a true eigenvalue of 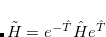 . Therefore we take the zero order Hamiltonian,
. Therefore we take the zero order Hamiltonian, 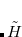 , to be the full in the p-space, while in the space of excluded substitutions (the q-space) we take only the one-body part of
, to be the full in the p-space, while in the space of excluded substitutions (the q-space) we take only the one-body part of 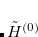 (which can be made diagonal). The fluctuation potential describing electron correlations in the
(which can be made diagonal). The fluctuation potential describing electron correlations in the 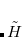 -space is
-space is  , and the (2) correction then follows from second-order perturbation theory.
, and the (2) correction then follows from second-order perturbation theory.
The new partitioning of terms between the perturbed and unperturbed Hamiltonians inherent in the (2) correction leads to a correction that shows both similarities and differences relative to the existing (T) corrections. There are two types of higher correlations that enter at second-order: not only triple substitutions, but also quadruple substitutions. The quadruples are treated with a factorization ansatz, that is exact in 5th order Møller-Plesset theory,[Kucharski and Bartlett(1998)] to reduce their computational cost from 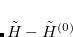 to
to 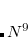 . For large basis sets this can still be larger than the cost of the triples terms, which scale as the 7th power of molecule size, with a factor twice as large as the usual (T) corrections.
. For large basis sets this can still be larger than the cost of the triples terms, which scale as the 7th power of molecule size, with a factor twice as large as the usual (T) corrections.
These corrections are feasible for molecules containing between four and ten first row atoms, depending on computer resources, and the size of the basis set chosen. There is early evidence that the (2) corrections are superior to the (T) corrections for highly correlated systems.[Gwaltney and Head-Gordon(2000)] This shows up in improved potential curves, particularly at long range and may also extend to improved energetic and structural properties at equilibrium in problematical cases. It will be some time before sufficient testing on the new (2) corrections has been done to permit a general assessment of the performance of these methods. However, they are clearly very promising, and for this reason they are available in Q-Chem.
6.9.3 (dT) and (fT) corrections
Alternative inclusion of non-iterative 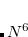 triples corrections is described in Section 7.7.21. These methods called (dT) and (fT) are of similar accuracy to other triples corrections. CCSD(dT) and CCSD(fT) are equivalent to the CR-CCSD(T)
triples corrections is described in Section 7.7.21. These methods called (dT) and (fT) are of similar accuracy to other triples corrections. CCSD(dT) and CCSD(fT) are equivalent to the CR-CCSD(T) and CR-CCSD(T)
and CR-CCSD(T) methods of Piecuch and coworkers.[Piecuch and Włoch(2005)]
methods of Piecuch and coworkers.[Piecuch and Włoch(2005)]
Note: Due to a violation of orbital invariance, the (dT) correction can sometimes lead to spurious results. Therefore, its use is discouraged. Use (fT) instead!
6.9.4 Job Control Options
The evaluation of a non-iterative (T) or (2) correction after a coupled-cluster singles and doubles level calculation (either CCSD, QCISD or OD) is controlled by the correlation keyword, and the specification of any frozen orbitals via N_FROZEN_CORE (and possibly N_FROZEN_VIRTUAL).
For the (2) correction, it is possible to apply the frozen core approximation in the reference coupled cluster calculation, and then correlate all orbitals in the (2) correction. This is controlled by CC_INCL_CORE_CORR, described below.
The default is to include core and core-valence correlation automatically in the CCSD(2) or OD(2) correction, if the reference CCSD or OD calculation was performed with frozen core orbitals. The reason for this choice is that core correlation is economical to include via this method (the main cost increase is only linear in the number of core orbitals), and such effects are important to account for in accurate calculations. This option should be made false if a job with explicitly frozen core orbitals is desired. One good reason for freezing core orbitals in the correction is if the basis set is physically inappropriate for describing core correlation (e.g., standard Pople basis sets, and Dunning cc-pV Z basis sets are designed to describe valence-only correlation effects). Another good reason is if a direct comparison is desired against another method such as CCSD(T) which is always used in the same orbital window as the CCSD reference.
Z basis sets are designed to describe valence-only correlation effects). Another good reason is if a direct comparison is desired against another method such as CCSD(T) which is always used in the same orbital window as the CCSD reference.
There are several implementations of non-iterative triples available in Q-Chem. In the original CCMAN suite, (T), (2), and (dT)/(fT) corrections can be computed. The parallel scaling of this code is very modest (4 cores max). CCMAN2 currently allows only the calculation of (T) correction for CCSD wave fucntions. By default, the CCMAN2 code is used for (T). The CCMAN code CCMAN2 is set to false. There are two versions of (T) in CCMAN2: The default version (native CCMAN2) and a new version using libpt. The implementation based on libpt is in-core MPI/OpenMP distributed-parallel. It is significantly faster in most realistic calculations (but it does not use point group symmetry, so it might show slower performance for small jobs with high symmetry). The libpt code is enabled by setting USE_LIBPT to true.
Note: For the best performance of libpt (T) code, parallel execution should be requested, see Section 2.8.
USE_LIBPT
Enable libpt for CCSD(T) calculations in CCMAN2.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
FALSE
RECOMMENDATION:
libpt is now used by default in all real-valued CC/EOM-CC calculations
CC_INCL_CORE_CORR
Whether to include the correlation contribution from frozen core orbitals in non iterative (2) corrections, such as OD(2) and CCSD(2).
TYPE:
LOGICAL
DEFAULT:
TRUE
OPTIONS:
TRUE
FALSE
RECOMMENDATION:
Use the default unless no core-valence or core correlation is desired (e.g., for comparison with other methods or because the basis used cannot describe core correlation).
6.9.5 Examples
Example 6.88 Two jobs that compare the correlation energy calculated via the standard CCSD(T) method with the new CCSD(2) approximation, both using the frozen core approximation. This requires that CC_INCL_CORE_CORR must be specified as FALSE in the CCSD(2) input.
$molecule
0 2
O
H O 0.97907
$end
$rem
METHOD ccsd(t)
BASIS cc-pvtz
N_FROZEN_CORE fc
$end
@@@
$molecule
read
$end
$rem
METHOD ccsd(2)
BASIS cc-pvtz
N_FROZEN_CORE fc
CC_INCL_CORE_CORR false
$end
Example 6.89 Using libpt for a standard CCSD(T) calculation
$molecule
0 2
O
H O 0.97907
$end
$rem
METHOD ccsd(t)
BASIS cc-pvtz
N_FROZEN_CORE fc
USE_LIBPT true
$end
Example 6.90 Water: Ground state CCSD(dT) calculation using RI
$molecule
0 1
O
H1 O OH
H2 O OH H1 HOH
OH = 0.957
HOH = 104.5
$end
$rem
JOBTYPE SP
BASIS cc-pvtz
AUX_BASIS rimp2-cc-pvtz
METHOD CCSD(dT)
$end