10.16 Linear–Scaling Computation of Electric Properties
The search for new optical devices is a major field of materials sciences. Here, polarizabilities and hyperpolarizabilities provide particularly important information on molecular systems. The response of the molecular systems in the presence of an external monochromatic oscillatory electric field is determined by the solution of the TDSCF equations, where the perturbation is represented as the interaction of the molecule with a single Fourier component within the dipole approximation:
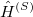 |
 |
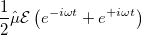 |
(10.66) | ||
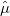 |
 |
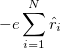 |
(10.67) |
Here, 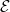 is the E-field vector,
is the E-field vector,  the corresponding frequency,
the corresponding frequency,  the electronic charge and
the electronic charge and 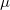 the dipole moment operator. Starting from Frenkel’s variational principle the TDSCF equations can be derived by standard techniques of perturbation theory [698]. As a solution we yield the first- [
the dipole moment operator. Starting from Frenkel’s variational principle the TDSCF equations can be derived by standard techniques of perturbation theory [698]. As a solution we yield the first- [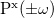 and second-order [e.g.,
and second-order [e.g., 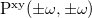 ] perturbed density matrices with which the following properties are calculated:
] perturbed density matrices with which the following properties are calculated:
Static polarizability:
![$\alpha _{\ensuremath{\mathrm{xy}}}(0;0) = \ensuremath{\mathrm{tr}}\left[\ensuremath{\mathbf{H}}^{\mu _{\ensuremath{\mathrm{x}}}}\ensuremath{\mathbf{P}}^{\ensuremath{\mathrm{y}}}\left(\omega =0\right)\right]$](images/img-1638.png)
Dynamic polarizability:
![$\alpha _{\ensuremath{\mathrm{xy}}}(\pm \omega ;\mp \omega ) = \ensuremath{\mathrm{tr}}\left[\ensuremath{\mathbf{H}}^{\mu _{\ensuremath{\mathrm{x}}}}\ensuremath{\mathbf{P}}^{\ensuremath{\mathrm{y}}}(\pm \omega )\right]$](images/img-1639.png)
Static hyperpolarizability:
![$\beta _{\ensuremath{\mathrm{xyz}}}(0;0,0) = \ensuremath{\mathrm{tr}}\left[\ensuremath{\mathbf{H}}^{\mu _{\ensuremath{\mathrm{x}}}}\ensuremath{\mathbf{P}}^{\ensuremath{\mathrm{yz}}}(\omega =0,\omega =0)\right]$](images/img-1640.png)
Second harmonic generation:
![$\beta _{\ensuremath{\mathrm{xyz}}}(\mp 2\omega ;\pm \omega ,\pm \omega ) = \ensuremath{\mathrm{tr}}\left[\ensuremath{\mathbf{H}}^{\mu _{\ensuremath{\mathrm{x}}}}\ensuremath{\mathbf{P}}^{\ensuremath{\mathrm{yz}}}(\pm \omega ,\pm \omega )\right]$](images/img-1641.png)
Electro-optical Pockels effect:
![$\beta _{\ensuremath{\mathrm{xyz}}}(\mp \omega ;0,\pm \omega ) = \ensuremath{\mathrm{tr}}\left[\ensuremath{\mathbf{H}}^{\mu _{\ensuremath{\mathrm{x}}}}\ensuremath{\mathbf{P}}^{\ensuremath{\mathrm{yz}}}(\omega =0,\pm \omega )\right]$](images/img-1642.png)
Optical rectification:
![$\beta _{\ensuremath{\mathrm{xyz}}}(0;\pm \omega ,\mp \omega ) = \ensuremath{\mathrm{tr}}\left[\ensuremath{\mathbf{H}}^{\mu _{\ensuremath{\mathrm{x}}}}\ensuremath{\mathbf{P}}^{\ensuremath{\mathrm{yz}}}(\pm \omega ,\mp \omega )\right]$](images/img-1643.png)
where 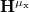 is the matrix representation of the
is the matrix representation of the  component of the dipole moments.
component of the dipole moments.
The TDSCF calculation is the most time consuming step and scales asymptotically as 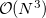 because of the AO/MO transformations. The scaling behavior of the two-electron integral formations, which dominate over a wide range because of a larger pre-factor, can be reduced by LinK/CFMM from quadratic to linear (
because of the AO/MO transformations. The scaling behavior of the two-electron integral formations, which dominate over a wide range because of a larger pre-factor, can be reduced by LinK/CFMM from quadratic to linear (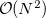
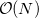 ).
).
Third-order properties can be calculated with the equations above after a second-order TDSCF calculation (MOPROP: 101/102) or by use of Wigner’s 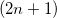 rule [699] (MOPROP: 103/104). Since the second order TDSCF depends on the first-order results, the convergence of the algorithm may be problematically. So we recommend the use of 103/104 for the calculation of first hyperpolarizabilities.
rule [699] (MOPROP: 103/104). Since the second order TDSCF depends on the first-order results, the convergence of the algorithm may be problematically. So we recommend the use of 103/104 for the calculation of first hyperpolarizabilities.
These optical properties can be computed for the first time using linear-scaling methods (LinK/CFMM) for all integral contractions [685]. Although the present implementation available in Q-Chem still uses MO-based time-dependent SCF (TDSCF) equations both at the HF and DFT level, the pre-factor of this 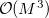 scaling step is rather small, so that the reduction of the scaling achieved for the integral contractions is most important. Here, all derivatives are computed analytically.
scaling step is rather small, so that the reduction of the scaling achieved for the integral contractions is most important. Here, all derivatives are computed analytically.
Further specifications of the dynamic properties are done in the section $fdpfreq in the following format:
$fdpfreq property frequencies units $end
The first line is only required for third order properties to specify the kind of first hyperpolarizability:
StaticHyper Static Hyperpolarizability
SHG Second harmonic generation
EOPockels Electro-optical Pockels effect
OptRect Optical rectification
Line number 2 contains floating-point values for the frequencies of the perturbations. Alternatively, for dynamic polarizabilities an equidistant sequence of frequencies can be specified by the keyword WALK (see example below). The last line specifies the units of the given frequencies:
au Frequency (atomic units)
eV Frequency (eV)
nm Wavelength (nm)
Hz Frequency (Hertz)
cmInv Wavenumber (
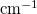 )
)
10.16.1 Examples for Section $fdpfreq
Example 10.255 Static and Dynamic polarizabilities, atomic units:
$fdpfreq
0.0 0.03 0.05
au
$end
Example 10.256 Series of dynamic polarizabilities, starting with 0.00 incremented by 0.01 up to 0.10:
$fdpfreq
walk 0.00 0.10 0.01
au
$end
Example 10.257 Static first hyperpolarizability, second harmonic generation and electro-optical Pockels effect, wavelength in nm:
$fdpfreq
StaticHyper SHG EOPockels
1064
nm
$end
10.16.2 Features of Mopropman
Restricted/unrestricted HF and KS-DFT CPSCF/TDSCF
LinK/CFMM support to evaluate Coulomb- and exchange-like matrices
DIIS acceleration
Support of LSDA/GGA/Hybrid XC functionals listed below
Analytical derivatives
The following XC functionals are supported:
Exchange:
Dirac
Becke 88
Correlation:
Wigner
VWN (both RPA and No. 5 parameterizations)
Perdew-Zunger 81
Perdew 86 (both PZ81 and VWN (No. 5) kernel)
LYP
10.16.3 Job Control
The following options can be used:
MOPROP
Specifies the job for mopropman.
TYPE:
INTEGER
DEFAULT:
0
Do not run mopropman.
OPTIONS:
1
NMR chemical shielding tensors.
2
Static polarizability.
3
Indirect nuclear spin–spin coupling tensors.
100
Dynamic polarizability.
101
First hyperpolarizability.
102
First hyperpolarizability, reading First order results from disk.
103
First hyperpolarizability using Wigner’s
104
First hyperpolarizability using Wigner’s
first order results from disk.
RECOMMENDATION:
None
MOPROP_PERTNUM
Set the number of perturbed densities that will to be treated together.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
All at once.
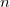
Treat the perturbed densities batch-wise.
RECOMMENDATION:
Use the default. For large systems, limiting this number may be required to avoid memory exhaustion.
MOPROP_CONV_1ST
Sets the convergence criteria for CPSCF and 1st order TDSCF.
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
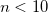
Convergence threshold set to
.
RECOMMENDATION:
None
MOPROP_CONV_2ND
Sets the convergence criterion for second-order TDSCF.
TYPE:
INTEGER
DEFAULT:
6
OPTIONS:
Convergence threshold set to
RECOMMENDATION:
None
MOPROP_MAXITER_1ST
The maximum number of iterations for CPSCF and first-order TDSCF.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
Set maximum number of iterations to
RECOMMENDATION:
Use the default.
MOPROP_MAXITER_2ND
The maximum number of iterations for second-order TDSCF.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
Set maximum number of iterations to
RECOMMENDATION:
Use the default.
MOPROP_ISSC_PRINT_REDUCED
Specifies whether the isotope-independent reduced coupling tensor
should be printed in addition to the isotope-dependent
-tensor when calculating indirect nuclear spin–spin couplings.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not print
TRUE
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_FC
Specifies whether to skip the calculation of the Fermi contact contribution to the indirect nuclear spin–spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate Fermi contact contribution.
TRUE
Skip Fermi contact contribution.
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_SD
Specifies whether to skip the calculation of the spin–dipole contribution to the indirect nuclear spin–spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate spin–dipole contribution.
TRUE
Skip spin–dipole contribution.
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_PSO
Specifies whether to skip the calculation of the paramagnetic spin–orbit contribution to the indirect nuclear spin–spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate paramagnetic spin–orbit contribution.
TRUE
Skip paramagnetic spin–orbit contribution.
RECOMMENDATION:
None
MOPROP_ISSC_SKIP_DSO
Specifies whether to skip the calculation of the diamagnetic spin–orbit contribution to the indirect nuclear spin–spin coupling tensor.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Calculate diamagnetic spin–orbit contribution.
TRUE
Skip diamagnetic spin–orbit contribution.
RECOMMENDATION:
None
MOPROP_DIIS
Controls the use of Pulay’s DIIS in solving the CPSCF equations.
TYPE:
INTEGER
DEFAULT:
5
OPTIONS:
0
Turn off DIIS.
5
Turn on DIIS.
RECOMMENDATION:
None
MOPROP_DIIS_DIM_SS
Specified the DIIS subspace dimension.
TYPE:
INTEGER
DEFAULT:
20
OPTIONS:
0
No DIIS.
Use a subspace of dimension
RECOMMENDATION:
None
SAVE_LAST_GPX
Save last
when calculating dynamic polarizabilities in order to call mopropman in a second run with MOPROP = 102.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
False
1
True
RECOMMENDATION:
None