9.3 Built-In ECPs
9.3.1 Overview
Q-Chem is equipped with several standard ECP sets which are specified using the ECP keyword within the $rem block. The built-in ECPs, which are described in some detail at the end of this Chapter, fall into four families:
The Hay-Wadt (or Los Alamos) sets (fit-HWMB and fit-LANL2DZ)
The Stevens-Basch-Krauss-Jansien-Cundari set (fit-SBKJC)
The Christiansen-Ross-Ermler-Nash-Bursten sets (fit-CRENBS and fit-CRENBL)
The Stuttgart-Bonn sets (SRLC and SRSC)
Besides the ones above, a common “def2-ECP" needs to be used with Karlsruhe basis sets for elements Rb-Rn (see section 8.3).
References and information about the definition and characteristics of most of these sets can be found at the EMSL site of the Pacific Northwest National Laboratory:[EMS()]
Each of the built-in ECPs comes with a matching orbital basis set for the valence electrons. In general, it is advisable to use these together and, if you select a basis set other than the matching one, Q-Chem will print a warning message in the output file. If you omit the BASIS $rem keyword entirely, Q-Chem will automatically provide the matching one.
The following $rem variable controls which ECP is used:
ECP
Defines the effective core potential and associated basis set to be used
TYPE:
STRING
DEFAULT:
No ECP
OPTIONS:
General, Gen
User defined. ($ecp keyword required)
Symbol
Use standard ECPs discussed above.
RECOMMENDATION:
ECPs are recommended for first row transition metals and heavier elements. Consul the reviews for more details.
9.3.2 Combining ECPs
If you wish, you can use different ECP sets for different elements in the system. This is especially useful if you would like to use a particular ECP but find that it is not available for all of the elements in your molecule. To combine different ECP sets, you set the ECP and BASIS keywords to “GEN” or (equivalently) “GENERAL”, and then add a $ecp block and a $basis block to your input file. In each of these blocks, you must name the ECP and the orbital basis set that you wish to use, separating each element by “****”. There is also a built-in combination that can be invoked specifying ECP = fit-LACVP. It assigns automatically 6-31G* or other suitable type basis sets for atoms H–Ar, while uses fit-LANL2DZ for heavier atoms.
9.3.3 Examples
Example 9.203 Computing the HF/fit-LANL2DZ energy of AgCl at a bond length of 2.4 Å.
$molecule
0 1
Ag
Cl Ag r
r = 2.4
$end
$rem
METHOD hf Hartree-Fock calculation
ECP fit-lanl2dz Using the Hay-Wadt ECP
BASIS lanl2dz And the matching basis set
$end
Example 9.204 Computing the single point energy of HI with B3LYP/def2-SV(P) (using def2-ECP for I).
$molecule
0 1
H 0.0 0.0 0.0
I 0.0 0.0 1.5
$end
$rem
METHOD b3lyp
BASIS def2-sv(p)
ECP def2-ecp
SYMMETRY false
SYM_IGNORE true
THRESH 14
SCF_CONVERGENCE 8
$end
Example 9.205 Optimization of the structure of Se using HF/fit-LANL2DZ, followed by a single-point energy calculation at the MP2/fit-LANL2DZ level.
using HF/fit-LANL2DZ, followed by a single-point energy calculation at the MP2/fit-LANL2DZ level.
$molecule
0 1
x1
x2 x1 xx
Se1 x1 sx x2 90.
Se2 x1 sx x2 90. Se1 90.
Se3 x1 sx x2 90. Se2 90.
Se4 x1 sx x2 90. Se3 90.
Se5 x2 sx x1 90. Se1 45.
Se6 x2 sx x1 90. Se5 90.
Se7 x2 sx x1 90. Se6 90.
Se8 x2 sx x1 90. Se7 90.
xx = 1.2
sx = 2.8
$end
$rem
JOBTYPE opt
METHOD hf
ECP fit-lanl2dz
$end
@@@
$molecule
read
$end
$rem
METHOD mp2 MP2 correlation energy
ECP fit-lanl2dz Hay-Wadt ECP and basis
SCF_GUESS read Read in the MOs
$end
Example 9.206 Computing the HF geometry of CdBr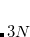 using the Stuttgart relativistic ECPs. The small-core ECP and basis are employed on the Cd atom and the large-core ECP and basis on the Br atoms.
using the Stuttgart relativistic ECPs. The small-core ECP and basis are employed on the Cd atom and the large-core ECP and basis on the Br atoms.
$molecule
0 1
Cd
Br1 Cd r
Br2 Cd r Br1 180.0
r = 2.4
$end
$rem
JOBTYPE opt Geometry optimization
METHOD hf Hartree-Fock theory
ECP gen Combine ECPs
BASIS gen Combine basis sets
PURECART 1 Use pure d functions
$end
$ecp
Cd
srsc
****
Br
srlc
****
$end
$basis
Cd
srsc
****
Br
srlc
****
$end