5.2 Treatment and the Definition of Core Electrons
Treatment of core electrons is controlled by N_FROZEN_CORE. Starting from version Q-Chem 5.0, the core electrons are frozen by default in most post-Hartree–Fock calculations. Selected virtual orbitals can also be frozen by using N_FROZEN_VIRTUAL keyword (the default for this is zero).
The number of core electrons in an atom is relatively well-defined, and consists of certain atomic shells. (Note that ECPs are available in both “small-core” and “large-core” varieties; see Chapter 8.) For example, in phosphorus the core consists of 1 , 2, and 2
, 2, and 2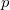 shells, for a total of ten electrons. In molecular systems, the core electrons are usually chosen as those occupying the
shells, for a total of ten electrons. In molecular systems, the core electrons are usually chosen as those occupying the 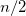 lowest energy orbitals, where
lowest energy orbitals, where 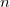 is the number of core electrons in the constituent atoms. In some cases, particularly in the lower parts of the periodic table, this definition is inappropriate and can lead to significant errors in the correlation energy. Vitaly Rassolov has implemented an alternative definition of core electrons within Q-Chem which is based on a Mulliken population analysis, and which addresses this problem [314].
is the number of core electrons in the constituent atoms. In some cases, particularly in the lower parts of the periodic table, this definition is inappropriate and can lead to significant errors in the correlation energy. Vitaly Rassolov has implemented an alternative definition of core electrons within Q-Chem which is based on a Mulliken population analysis, and which addresses this problem [314].
The current implementation is restricted to n-kl type basis sets such as 3-21 or 6-31, and related bases such as 6-31+G(d). There are essentially two cases to consider, the outermost 6G functions (or 3G in the case of the 3-21G basis set) for Na, Mg, K and Ca, and the 3d functions for the elements Ga—Kr. Whether or not these are treated as core or valence is determined by the CORE_CHARACTER $rem, as summarized in Table 5.2.
CORE_CHARACTER |
Outermost 6G (3G) |
3d (Ga–Kr) |
for Na, Mg, K, Ca |
||
1 |
valence |
valence |
2 |
valence |
core |
3 |
core |
core |
4 |
core |
valence |
N_FROZEN_CORE
Sets the number of frozen core orbitals in a post-Hartree–Fock calculation.
TYPE:
INTEGER
DEFAULT:
FC
OPTIONS:
FC
Frozen Core approximation (all core orbitals frozen).
Freeze
RECOMMENDATION:
Correlated calculations calculations are more efficient with frozen core orbitals. Use default if possible.
N_FROZEN_VIRTUAL
Sets the number of frozen virtual orbitals in a post-Hartree–Fock calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
Freeze
RECOMMENDATION:
None
CORE_CHARACTER
Selects how the core orbitals are determined in the frozen-core approximation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use energy-based definition.
1-4
Use Mulliken-based definition (see Table 5.2 for details).
RECOMMENDATION:
Use the default, unless performing calculations on molecules with heavy elements.
PRINT_CORE_CHARACTER
Determines the print level for the CORE_CHARACTER option.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No additional output is printed.
1
Prints core characters of occupied MOs.
2
Print level 1, plus prints the core character of AOs.
RECOMMENDATION:
Use the default, unless you are uncertain about what the core character is.