8.4 User-Defined Basis Sets ($basis)
8.4.1 Introduction
Users may, on occasion, prefer to use non-standard basis, and it is possible to declare user-defined basis sets in Q-Chem input (see Chapter 3 on Q-Chem inputs). The format for inserting a non-standard user-defined basis set is both logical and flexible, and is described in detail in the job control section below.
Note that the SAD guess is not currently supported with non-standard or user-defined basis sets. The simplest alternative is to specify the GWH or CORE options for SCF_GUESS, but these are relatively ineffective other than for small basis sets. The recommended alternative is to employ basis set projection by specifying a standard basis set for the BASIS2 keyword. See the section in Chapter 4 on initial guesses for more information.
8.4.2 Job Control
In order to use a user-defined basis set the BASIS $rem must be set to GENERAL or GEN.
When using a non-standard basis set which incorporates  or higher angular momentum basis functions, the $rem variable PURECART needs to be initiated. This $rem variable indicates to the Q-Chem program how to handle the angular form of the basis functions. As indicated above, each integer represents an angular momentum type which can be defined as either pure (1) or Cartesian (2). For example, 111 would specify all
or higher angular momentum basis functions, the $rem variable PURECART needs to be initiated. This $rem variable indicates to the Q-Chem program how to handle the angular form of the basis functions. As indicated above, each integer represents an angular momentum type which can be defined as either pure (1) or Cartesian (2). For example, 111 would specify all  ,
, 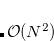 and basis functions as being in the pure form. 121 would indicate - and - functions are pure and -functions Cartesian.
and basis functions as being in the pure form. 121 would indicate - and - functions are pure and -functions Cartesian.
PURECART
INTEGER
TYPE:
Controls the use of pure (spherical harmonic) or Cartesian angular forms
DEFAULT:
2111
Cartesian
-functions and pure
functions
OPTIONS:

Use 1 for pure and 2 for Cartesian.
RECOMMENDATION:
This is pre-defined for all standard basis sets
In standard basis sets all functions are pure, except for the functions in  -21G–type bases (e.g., 3-21G) and -31G bases (e.g., 6-31G, 6-31G*,6-31+G*,
-21G–type bases (e.g., 3-21G) and -31G bases (e.g., 6-31G, 6-31G*,6-31+G*, 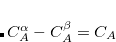 ). In particular, the 6-311G series uses pure functions for both and .
). In particular, the 6-311G series uses pure functions for both and .
8.4.3 Format for User-Defined Basis Sets
The format for the user-defined basis section is as follows: $basis 0 **** $end

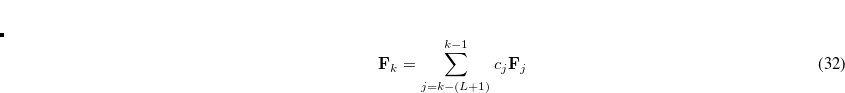















where
|
Atomic symbol of the atom (atomic number not accepted) |
|
Angular momentum symbol (S, P, SP, D, F, G) |
|
Degree of contraction of the shell (integer) |
|
Scaling to be applied to exponents (default is 1.00) |
|
Gaussian primitive exponent (positive real number) |
|
Contraction coefficient for each angular momentum (non-zero real numbers). |


Atoms are terminated with **** and the complete basis set is terminated with the $end keyword terminator. No blank lines can be incorporated within the general basis set input. Note that more than one contraction coefficient per line is one required for compound shells like SP. As with all Q-Chem input deck information, all input is case-insensitive.
8.4.4 Example
Example 8.198 Example of adding a user-defined non-standard basis set. Note that since , and functions are incorporated, the $rem variable PURECART must be set. Note the use of BASIS2 for the initial guess.
$molecule
0 1
O
H O oh
H O oh 2 hoh
oh = 1.2
hoh = 110.0
$end
$rem
EXCHANGE hf
BASIS gen user-defined general basis
BASIS2 sto-3g sto-3g orbitals as initial guess
PURECART 112 Cartesian d functions, pure f and g
$end
$basis
H 0
S 2 1.00
1.30976 0.430129
0.233136 0.678914
****
O 0
S 2 1.00
49.9810 0.430129
8.89659 0.678914
SP 2 1.00
1.94524 0.049472 0.511541
0.49336 0.963782 0.612820
D 1 1.00
0.39000 1.000000
F 1 1.00
4.10000 1.000000
G 1 1.00
3.35000 1.000000
****
$end