10.7 Spin and Charge Densities at the Nuclei
Gaussian basis sets violate nuclear cusp conditions [626, 627, 628]. This may lead to large errors in wave function at nuclei, particularly for spin density calculations [629]. This problem can be alleviated by using an averaging operator that compute wave function density based on constraints that wave function must satisfy near Coulomb singularity [630, 631]. The derivation of operators is based on hyper virial theorem [632] and presented in Ref. Rassolov:1996b. Application to molecular spin densities for spin-polarized [631] and DFT [633] wave functions show considerable improvement over traditional delta function operator.
One of the simplest forms of such operators is based on the Gaussian weight function ![$\exp [-(Z/r_{0})^{2}(\ensuremath{\mathbf{r}}-\ensuremath{\mathbf{R}})^{2}]$](images/img-1491.png) that samples the vicinity of a nucleus of charge
that samples the vicinity of a nucleus of charge 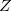 located at
located at 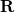 . The parameter
. The parameter 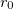 has to be small enough to neglect two-electron contributions of the order
has to be small enough to neglect two-electron contributions of the order 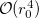 but large enough for meaningful averaging. The range of values between 0.15–0.3 a.u. has been shown to be adequate, with final answer being relatively insensitive to the exact choice of [630, 631]. The value of is chosen by RC_R0 keyword in the units of 0.001 a.u. The averaging operators are implemented for single determinant Hartree-Fock and DFT, and correlated SSG wave functions. Spin and charge densities are printed for all nuclei in a molecule, including ghost atoms.
but large enough for meaningful averaging. The range of values between 0.15–0.3 a.u. has been shown to be adequate, with final answer being relatively insensitive to the exact choice of [630, 631]. The value of is chosen by RC_R0 keyword in the units of 0.001 a.u. The averaging operators are implemented for single determinant Hartree-Fock and DFT, and correlated SSG wave functions. Spin and charge densities are printed for all nuclei in a molecule, including ghost atoms.
RC_R0
Determines the parameter in the Gaussian weight function used to smooth the density at the nuclei.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Corresponds the traditional delta function spin and charge densities
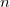
corresponding to
a.u.
RECOMMENDATION:
We recommend value of 250 for a typical spit valence basis. For basis sets with increased flexibility in the nuclear vicinity the smaller values of