12.12 Energy Decomposition Analysis based on SAPT/cDFT
Many schemes for decomposing quantum chemical calculations of intermolecular interaction energies into physically meaningful components can be found in the literature, but the definition of the charge-transfer (CT) contribution has proven particularly vexing to define in a satisfactory way and typically depends strongly on the choice of basis set,[882, 883, 853] because as virtual orbitals on monomer 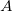 start to extend significantly over monomer
start to extend significantly over monomer 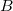 as the basis set approaches completeness, the distinction between polarization (excitations localized on , introduced by the perturbing influence of ) and CT (excitations from to ) becomes blurred [853]. This ambiguity renders orbital-dependent definitions of CT highly dependent on the choice of atomic orbital basis set. On the other hand, constrained density functional theory (cDFT, Section 4.11), where a CT-free reference state can be defined based on “promolecule” densities, affords a definition of CT that is scarcely dependent on the basis set and is in accord with chemical intuition in simple cases.[853]
as the basis set approaches completeness, the distinction between polarization (excitations localized on , introduced by the perturbing influence of ) and CT (excitations from to ) becomes blurred [853]. This ambiguity renders orbital-dependent definitions of CT highly dependent on the choice of atomic orbital basis set. On the other hand, constrained density functional theory (cDFT, Section 4.11), where a CT-free reference state can be defined based on “promolecule” densities, affords a definition of CT that is scarcely dependent on the basis set and is in accord with chemical intuition in simple cases.[853]
For intermolecular interactions, the cDFT definition of CT can be combined with a definition of the remaining components of the interaction energy (electrostatics, induction, Pauli repulsion, and van der Waals interactions) based on symmetry-adapted perturbation theory (SAPT, Section 12.10). In traditional SAPT, the CT interaction energy resides within the induction energy (also known as the polarization energy), which is therefore itself highly dependent upon the basis set. However, using cDFT to define the CT component and subtracting this out of the SAPT induction energy, both the CT and the remaining induction energies are largely independent of basis set [853]. SAPT/cDFT therefore provides a stable and physically-motivated energy decomposition, which can be invoked by setting the $rem variable SAPT_CDFT_EDA = TRUE in a SAPT calculation. A $cdft section must be set to specify the monomer charges and spins for the cDFT calculation. Researchers who use Q-Chem’s SAPT/cDFT code are asked to cite Ref. Lao:2016b.
SAPT_CDFT_EDA
Request a SAPT/cDFT energy decomposition analysis
TYPE:
BOOLEAN
DEFAULT:
FALSE
OPTIONS:
TRUE
Run a SAPT/cDFT calculation.
FALSE
Do not run SAPT/cDFT.
RECOMMENDATION:
None
Example 12.321 Energy decomposition analysis for the water dimer using AO-SAPT+D3/cDFT.
$comment
a $cdft section must be set to specify the monomer charges and spins
for the cDFT calculation.
$end
$rem
SYM_IGNORE true
EXCHANGE gen
BASIS aug-cc-pvdz
XPOL true ! must be set to true for sapt jobs too
XPOL_MPOL_ORDER gas ! gas or charges
XPOL_OMEGA true
XPOL_PRINT 3
SAPT_PRINT 3
SAPT true
SAPT_AO true
SAPT_ORDER 2 ! can be set to 1, ELST or RSPT
SAPT_BASIS dimer ! monomer, dimer (if only 2 monomers), or projected
SAPT_DISP_CORR true
SAPT_DISP_VERSION 3
LRC_DFT true
SAPT_CDFT_EDA true
$end
$xc_functional
x wPBE 1.0
c PBE 1.0
$end
$lrc_omega
500
500
$end
$cdft
0
1 1 3
0
1 1 3 s
$end
$molecule
0 1
--
0 1
O -0.702196054 -0.056060256 0.009942262
H -1.022193224 0.846775782 -0.011488714
H 0.257521062 0.042121496 0.005218999
--
0 1
O 2.220871067 0.026716792 0.000620476
H 2.597492682 -0.411663274 0.766744858
H 2.593135384 -0.449496183 -0.744782026
$end