6.5 Restricted Open-Shell Kohn-Sham Method for 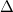 -SCF Calculations of Excited States
-SCF Calculations of Excited States
Q-Chem provides access to certain singlet excited states – namely, those well-described by a single-electron HOMO-LUMO transition – via restricted open-shell Kohn-Sham (ROKS) theory. In contrast to the MOM approach (see Section 6.4), which requires separate SCF calculations of the non-aufbau and triplet energies, the ROKS approach attempts to combine the properties of both determinants at the level of the Fock matrix in one SCF calculation. ROKS thus presents as a single SCF loop, but the structure of the Fock matrix differs from the ground-state case. Note that this excited-state method is distinct from ROKS theory for open-shell ground states.
The implementation of ROKS excited states in Q-Chem largely follows the theoretical framework established by Filatov and Shaik[436] and is described in detail in Ref. Kowalczyk:2013. Singlet excited state energies and gradients are available, enabling single-point, geometry optimization and molecular dynamics.
To perform an ROKS excited state calculation, simply set the keywords ROKS = TRUE and UNRESTRICTED = FALSE. An additional keyword ROKS_LEVEL_SHIFT is included to assist in cases of convergence difficulties with a standard level-shift technique. It is recommended to perform a preliminary ground-state calculation on the system first, and then use the ground-state orbitals to construct the initial guess using SCF_GUESS = READ.
ROKS
Controls whether ROKS calculation will be performed.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
ROKS is not performed.
TRUE
ROKS will be performed.
RECOMMENDATION:
Set to TRUE if ROKS calculation is desired. You should also set UNRESTRICTED = FALSE
ROKS_LEVEL_SHIFT
Introduce a level shift of N/100 hartree to aid convergence.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
No shift
N
level shift of N/100 hartree.
RECOMMENDATION:
Use in cases of problematic convergence.
Example 6.121 RO-PBE0/6-311+G* excited state gradient of formaldehyde, using the ground state orbitals as an initial guess.
$comment
ROKS excited state gradient of formaldehyde
Use orbitals from ground state for initial guess
$end
$rem
EXCHANGE pbe0
BASIS 6-311+G*
SCF_CONVERGENCE 9
SYM_IGNORE true
$end
$molecule
0 1
H -0.940372 0.000000 1.268098
H 0.940372 0.000000 1.268098
C 0.000000 0.000000 0.682557
O 0.000000 0.000000 -0.518752
$end
@@@
$molecule
read
$end
$rem
ROKS true
UNRESTRICTED false
EXCHANGE pbe0
BASIS 6-311+G*
JOBTYPE force
SCF_CONVERGENCE 9
SYM_IGNORE true
SCF_GUESS read
$end