9.1 Equilibrium Geometries and Transition-State Structures
9.1.1 Overview
Molecular potential energy surfaces rely on the Born-Oppenheimer separation of nuclear and electronic motion. Minima on such energy surfaces correspond to the classical picture of equilibrium geometries, and transition state structures correspond to first-order saddle points. Both equilibrium and transition-state structures are stationary points for which the energy gradient vanishes. Characterization of such critical points requires consideration of the eigenvalues of the Hessian (second derivative matrix): minimum-energy, equilibrium geometries possess Hessians whose eigenvalues are all positive, whereas transition-state structures are defined by a Hessian with precisely one negative eigenvalue. (The latter is therefore a local maximum along the reaction path between minimum-energy reactant and product structures, but a minimum in all directions perpendicular to this reaction path.
The quality of a geometry optimization algorithm is of major importance; even the fastest integral code in the world will be useless if combined with an inefficient optimization algorithm that requires excessive numbers of steps to converge. Q-Chem incorporates a geometry optimization package (Optimize—see Appendix A) developed by the late Jon Baker over more than ten years.
The key to optimizing a molecular geometry successfully is to proceed from the starting geometry to the final geometry in as few steps as possible. Four factors influence the path and number of steps:
starting geometry
optimization algorithm
quality of the Hessian (and gradient)
coordinate system
Q-Chem controls the last three of these, but the starting geometry is solely determined by the user, and the closer it is to the converged geometry, the fewer optimization steps will be required. Decisions regarding the optimization algorithm and the coordinate system are generally made by the Optimize package (i.e., internally, within Q-Chem) to maximize the rate of convergence. Although users may override these choices in many cases, this is not generally recommended.
Level of Theory |
Analytical |
Maximum Angular |
Analytical |
Maximum Angular |
(Algorithm) |
Gradients |
Momentum Type |
Hessian |
Momentum Type |
DFT |
✓ |
|
✓ |
|
HF |
✓ |
|
✓ |
|
ROHF |
✓ |
|
✗ |
|
MP2 |
✓ |
|
✗ |
|
(V)OD |
✓ |
|
✗ |
|
(V)QCCD |
✓ |
|
✗ |
|
CIS (except RO) |
✓ |
|
✓ |
|
CFMM |
✓ |
|
✗ |
Another consideration when trying to minimize the optimization time concerns the quality of the gradient and Hessian. A higher-quality Hessian (i.e., analytical versus approximate) will in many cases lead to faster convergence, in the sense of requiring fewer optimization steps. However, the construction of an analytical Hessian requires significant computational effort and may outweigh the advantage of fewer optimization cycles. Currently available analytical gradients and Hessians are summarized in Table 9.1.
Features of Q-Chem’s geometry and transition-state optimization capabilities include:
Cartesian, Z-matrix or internal coordinate systems
Eigenvector Following (EF) or GDIIS algorithms
Constrained optimizations
Equilibrium structure searches
Transition structure searches
Initial Hessian and Hessian update options
Reaction pathways using intrinsic reaction coordinates (IRC)
Optimization of minimum-energy crossing points (MECPs) along conical seams
9.1.2 Job Control
Obviously a level of theory, basis set, and starting molecular geometry must be specified to begin a geometry optimization or transition-structure search. These aspects are described elsewhere in this manual, and this section describes job-control variables specific to optimizations.
JOBTYPE
Specifies the calculation.
TYPE:
STRING
DEFAULT:
Default is single-point, which should be changed to one of the following options.
OPTIONS:
OPT
Equilibrium structure optimization.
TS
Transition structure optimization.
RPATH
Intrinsic reaction path following.
RECOMMENDATION:
Application-dependent.
GEOM_OPT_HESSIAN
Determines the initial Hessian status.
TYPE:
STRING
DEFAULT:
DIAGONAL
OPTIONS:
DIAGONAL
Set up diagonal Hessian.
READ
Have exact or initial Hessian. Use as is if Cartesian, or transform
if internals.
RECOMMENDATION:
An accurate initial Hessian will improve the performance of the optimizer, but is expensive to compute.
GEOM_OPT_COORDS
Controls the type of optimization coordinates.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
0
Optimize in Cartesian coordinates.
1
Generate and optimize in internal coordinates, if this fails abort.
Generate and optimize in internal coordinates, if this fails at any stage of the
optimization, switch to Cartesian and continue.
2
Optimize in
-matrix coordinates, if this fails abort.
Optimize in
optimization switch to Cartesians and continue.
RECOMMENDATION:
Use the default; delocalized internals are more efficient.
GEOM_OPT_TOL_GRADIENT
Convergence on maximum gradient component.
TYPE:
INTEGER
DEFAULT:
300
tolerance on maximum gradient component.
OPTIONS:
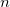
Integer value (tolerance =
).
RECOMMENDATION:
Use the default. To converge GEOM_OPT_TOL_GRADIENT and one of GEOM_OPT_TOL_DISPLACEMENT and GEOM_OPT_TOL_ENERGY must be satisfied.
GEOM_OPT_TOL_DISPLACEMENT
Convergence on maximum atomic displacement.
TYPE:
INTEGER
DEFAULT:
1200
tolerance on maximum atomic displacement.
OPTIONS:
Integer value (tolerance =
RECOMMENDATION:
Use the default. To converge GEOM_OPT_TOL_GRADIENT and one of GEOM_OPT_TOL_DISPLACEMENT and GEOM_OPT_TOL_ENERGY must be satisfied.
GEOM_OPT_TOL_ENERGY
Convergence on energy change of successive optimization cycles.
TYPE:
INTEGER
DEFAULT:
100
tolerance on maximum (absolute) energy change.
OPTIONS:
).
RECOMMENDATION:
Use the default. To converge GEOM_OPT_TOL_GRADIENT and one of GEOM_OPT_TOL_DISPLACEMENT and GEOM_OPT_TOL_ENERGY must be satisfied.
GEOM_OPT_MAX_CYCLES
Maximum number of optimization cycles.
TYPE:
INTEGER
DEFAULT:
50
OPTIONS:
User defined positive integer.
RECOMMENDATION:
The default should be sufficient for most cases. Increase if the initial guess geometry is poor, or for systems with shallow potential wells.
GEOM_OPT_PRINT
Controls the amount of Optimize print output.
TYPE:
INTEGER
DEFAULT:
3
Error messages, summary, warning, standard information and gradient print out.
OPTIONS:
0
Error messages only.
1
Level 0 plus summary and warning print out.
2
Level 1 plus standard information.
3
Level 2 plus gradient print out.
4
Level 3 plus Hessian print out.
5
Level 4 plus iterative print out.
6
Level 5 plus internal generation print out.
7
Debug print out.
RECOMMENDATION:
Use the default.
GEOM_OPT_SYMFLAG
Controls the use of symmetry in Optimize.
TYPE:
INTEGER
DEFAULT:
1
OPTIONS:
1
Make use of point group symmetry.
0
Do not make use of point group symmetry.
RECOMMENDATION:
Use the default.
GEOM_OPT_MODE
Determines Hessian mode followed during a transition state search.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Mode following off.
Maximize along mode
RECOMMENDATION:
Use the default, for geometry optimizations.
GEOM_OPT_MAX_DIIS
Controls maximum size of subspace for GDIIS.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Do not use GDIIS.
-1
Default size = min(NDEG, NATOMS, 4) NDEG = number of molecular
degrees of freedom.
Size specified by user.
RECOMMENDATION:
Use the default or do not set
GEOM_OPT_DMAX
Maximum allowed step size. Value supplied is multiplied by 10
.
TYPE:
INTEGER
DEFAULT:
300
= 0.3
OPTIONS:
User-defined cutoff.
RECOMMENDATION:
Use the default.
GEOM_OPT_UPDATE
Controls the Hessian update algorithm.
TYPE:
INTEGER
DEFAULT:
-1
OPTIONS:
-1
Use the default update algorithm.
0
Do not update the Hessian (not recommended).
1
Murtagh-Sargent update.
2
Powell update.
3
Powell/Murtagh-Sargent update (TS default).
4
BFGS update (OPT default).
5
BFGS with safeguards to ensure retention of positive definiteness
(GDISS default).
RECOMMENDATION:
Use the default.
GEOM_OPT_LINEAR_ANGLE
Threshold for near linear bond angles (degrees).
TYPE:
INTEGER
DEFAULT:
165 degrees.
OPTIONS:
User-defined level.
RECOMMENDATION:
Use the default.
FDIFF_STEPSIZE
Displacement used for calculating derivatives by finite difference.
TYPE:
INTEGER
DEFAULT:
100
Corresponding to 0.001 . For calculating second derivatives.
OPTIONS:
Use a step size of
.
RECOMMENDATION:
Use the default except in cases where the potential surface is very flat, in which case a larger value should be used. See FDIFF_STEPSIZE_QFF for third and fourth derivatives.
Example 9.191 As outlined, the rate of convergence of the iterative optimization process is dependent on a number of factors, one of which is the use of an initial analytic Hessian. This is easily achieved by instructing Q-Chem to calculate an analytic Hessian and proceed then to determine the required critical point
$molecule
0 1
O
H 1 oh
H 1 oh 2 hoh
oh = 1.1
hoh = 104
$end
$rem
JOBTYPE freq Calculate an analytic Hessian
METHOD hf
BASIS 6-31g(d)
$end
$comment
Now proceed with the Optimization making sure to read in the analytic
Hessian (use other available information too).
$end
@@@
$molecule
read
$end
$rem
JOBTYPE opt
METHOD hf
BASIS 6-31g(d)
SCF_GUESS read
GEOM_OPT_HESSIAN read Have the initial Hessian
$end