8.4 User-Defined ECPs
Many users will find that the library of built-in ECPs is adequate for their needs. However, if you need to use an ECP that is not built into Q-Chem, you can enter it in much the same way as you can enter a user-defined orbital basis set (see Chapter 7).
8.4.1 Job Control for User-Defined ECPs
To apply a user-defined ECP, you must set the ECP and BASIS keywords in $rem to “Gen”. You then add a $ecp block that defines your ECP, element by element, and a $basis block that defines your orbital basis set, separating elements by asterisks.
The syntax within the $basis block is described in Chapter 7. The syntax for each record within the $ecp block is as follows:.
$ecp
For each atom that will bear an ECP
Chemical symbol for the atom
ECP name ; the 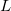 value for the ECP ; number of core electrons removed
value for the ECP ; number of core electrons removed
For each ECP component (in the order unprojected, 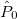 ,
, 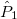 , ,
, , 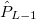
The component name
The number of Gaussians in the component
For each Gaussian in the component
The power of  ; the exponent ; the contraction coefficient
; the exponent ; the contraction coefficient
A sequence of four asterisks (i.e., ****)
$end
Note: (1) All of the information in the $ecp block is case-insensitive.
(2) The value may not exceed 4. That is, nothing beyond 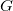 projectors is allowed.
projectors is allowed.
(3) The power of (which includes the Jacobian 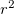 factor) must be 0, 1 or 2.
factor) must be 0, 1 or 2.
8.4.2 Example
Example 8.205 Optimizing the HF geometry of AlH using a user-defined ECP and basis set on Al and the 3-21G basis on H.
using a user-defined ECP and basis set on Al and the 3-21G basis on H.
$molecule
0 1
Al
H1 Al r
H2 Al r H1 120
H3 Al r H1 120 H2 180
r = 1.6
$end
$rem
JOBTYPE opt Geometry optimization
METHOD hf Hartree-Fock theory
ECP gen User-defined ECP
BASIS gen User-defined basis
$end
$ecp
Al
Stevens_ECP 2 10
d potential
1
1 1.95559 -3.03055
s-d potential
2
0 7.78858 6.04650
2 1.99025 18.87509
p-d potential
2
0 2.83146 3.29465
2 1.38479 6.87029
****
$end
$basis
Al
SP 3 1.00
0.90110 -0.30377 -0.07929
0.44950 0.13382 0.16540
0.14050 0.76037 0.53015
SP 1 1.00
0.04874 0.32232 0.47724
****
H
3-21G
****
$end