12.6 Job Control for Locally-Projected SCF Methods
FRGM_METHOD
Specifies a locally-projected method.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
STOLL
Locally-projected SCF equations of Stoll are solved.
GIA
Locally-projected SCF equations of Gianinetti are solved.
NOSCF_RS
Single Roothaan-step correction to the FRAGMO initial guess.
NOSCF_ARS
Approximate single Roothaan-step correction to the FRAGMO initial guess.
NOSCF_DRS
Double Roothaan-step correction to the FRAGMO initial guess.
NOSCF_RS_FOCK
Non-converged SCF energy of the single Roothaan-step MOs.
RECOMMENDATION:
STOLL and GIA are for variational optimization of the ALMOs. NOSCF options are for computationally fast corrections of the FRAGMO initial guess.
FRGM_LPCORR
Specifies a correction method performed after the locally-projected equations are converged.
TYPE:
STRING
DEFAULT:
NONE
OPTIONS:
ARS
Approximate Roothaan-step perturbative correction.
RS
Single Roothaan-step perturbative correction.
EXACT_SCF
Full SCF variational correction.
ARS_EXACT_SCF
Both ARS and EXACT_SCF in a single job.
RS_EXACT_SCF
Both RS and EXACT_SCF in a single job.
RECOMMENDATION:
For large basis sets use ARS, use RS if ARS fails.
SCF_PRINT_FRGM
Controls the output of Q-Chem jobs on isolated fragments.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE
The output is printed to the parent job output file.
FALSE
The output is not printed.
RECOMMENDATION:
Use TRUE if details about isolated fragments are important.
EDA_BSSE
Calculates the BSSE correction when performing the energy decomposition analysis.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set to TRUE unless a very large basis set is used.
EDA_COVP
Perform COVP analysis when evaluating the RS or ARS charge-transfer correction. COVP analysis is currently implemented only for systems of two fragments.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set to TRUE to perform COVP analysis in an EDA or SCF MI(RS) job.
EDA_PRINT_COVP
Replace the final MOs with the CVOP orbitals in the end of the run.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
TRUE/FALSE
RECOMMENDATION:
Set to TRUE to print COVP orbitals instead of conventional MOs.
NVO_LIN_MAX_ITE
Maximum number of iterations in the preconditioned conjugate gradient solver of the single-excitation amplitude equations.
TYPE:
INTEGER
DEFAULT:
30
OPTIONS:
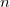
User–defined number of iterations.
RECOMMENDATION:
None.
NVO_LIN_CONVERGENCE
Target error factor in the preconditioned conjugate gradient solver of the single-excitation amplitude equations.
TYPE:
INTEGER
DEFAULT:
3
OPTIONS:
User–defined number.
RECOMMENDATION:
Solution of the single-excitation amplitude equations is considered converged if the maximum residual is less than
multiplied by the current DIIS error. For the ARS correction,
NVO_METHOD
Sets method to be used to converge solution of the single-excitation amplitude equations.
TYPE:
INTEGER
DEFAULT:
9
OPTIONS:
User–defined number.
RECOMMENDATION:
This is an experimental option. Use the default.
NVO_UVV_PRECISION
Controls convergence of the Taylor series when calculating the
block from the single-excitation amplitudes. Series is considered converged when the maximum element of the term is less than
TYPE:
INTEGER
DEFAULT:
11
OPTIONS:
User–defined number.
RECOMMENDATION:
NVO_UVV_PRECISION must be the same as or larger than THRESH.
NVO_UVV_MAXPWR
Controls convergence of the Taylor series when calculating the
TYPE:
INTEGER
DEFAULT:
10
OPTIONS:
User–defined number.
RECOMMENDATION:
None.
NVO_TRUNCATE_DIST
Specifies which atomic blocks of the Fock matrix are used to construct the preconditioner.
TYPE:
INTEGER
DEFAULT:
-1
OPTIONS:
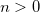
If distance between a pair of atoms is more than
do not include the atomic block.
-2
Do not use distance threshold, use NVO_TRUNCATE_PRECOND instead.
-1
Include all blocks.
0
Include diagonal blocks only.
RECOMMENDATION:
This option does not affect the final result. However, it affects the rate of the PCG algorithm convergence. For small systems, use the default.
NVO_TRUNCATE_PRECOND
Specifies which atomic blocks of the Fock matrix are used to construct the preconditioner. This variable is used only if NVO_TRUNCATE_DIST is set to
.
TYPE:
INTEGER
DEFAULT:
2
OPTIONS:
If the maximum element in an atomic block is less than
the block.
RECOMMENDATION:
Use the default. Increasing