12.2 Specifying Fragments in the $molecule Section
To request any of the methods mentioned above one must specify how system is partitioned into fragments. All atoms and all electrons in the systems should be assigned to a fragment. Each fragment must contain an integer number of electrons. In the current implementation, both open and closed-shell fragments are allowed. In order to specify fragments, the fragment descriptors must be inserted into the $molecule section of the Q-Chem input file. A fragment descriptor consists of two lines: the first line must start with two hyphens followed by optional comments, the second line must contain the charge and the multiplicity of the fragment. At least two fragments must be specified. Fragment descriptors in the $molecule section does not affect jobs that are not designed to use fragmentation.
Example 12.298 Fragment descriptors in the $molecule section.
$molecule
0 1
-- water molecule - proton donor
0 1
O1
H2 O1 0.96
H3 O1 0.96 H2 105.4
-- water molecule - proton acceptor
0 1
O4 O1 ROO H2 105.4 H3 0.0
X5 O4 2.00 O1 120.0 H2 180.0
H6 O4 0.96 X5 55.6 O1 90.0
H7 O4 0.96 X5 55.6 01 -90.0
ROO = 2.4
$end
Open shell systems must have a number of alpha electrons greater than the number of beta electrons. However, individual fragments in the system can be made to contain excess beta electrons by specifying a negative multiplicity. For instance, a multiplicity of 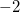 indicates one excess beta electron, as in the second fragment of the following example.
indicates one excess beta electron, as in the second fragment of the following example.
Example 12.299 Open shell fragment descriptors in the $molecule section. The second fraghment is made with a negative multiplicity, so that overall the number of alpha and beta electrons match, yielding an approximate singlet state.
$molecule
0 1
-- An alpha spin H atom
0 2
H1
-- A beta spin H atom
0 -2
H2 H1 1.50
$end