10.4 Fewest-switches surface hopping
The fewest switches surface hopping (FSSH) [518] non-adiabatic dynamics method is available for any electronic structure method for which derivative couplings (Section 10.3) are available. The nuclear dynamics component of the simulation can be specified by the same variables used to request an AIMD calculation (Section 9.9). Artificial decoherence can be added to the calculation at additional cost according to the augmented FSSH (AFSSH) method, [519, 520, 521] which enforces stochastic wave function collapse at a rate proportional to the difference in forces between the trajectory on the active surface and position moments propagated the other surfaces.
At every time step, the component of the wave function on each active surface is printed to the output file. These amplitudes, as well as the position and momentum moments (if AFSSH is requested), is also printed to a text file called “SurfaceHopper” located in the “AIMD” directory, which is a subdirectory of the job’s scratch directory.
In order to request a FSSH calculation, only a few aditional $rem variables must be added to those necessary for an excited state AIMD input file. Currently, FSSH calculations can only be performed using Born-Oppenheimer molecular dynamics (BOMD) method. Furthermore, the optimized velocity Verlet (OVV) integration method is not supported for FSSH calculations.
10.4.1 Job Control
FSSH_LOWESTSURFACE
Specifies the lowest-energy state considered in a surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
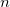
Only states
RECOMMENDATION:
None
FSSH_NSURFACES
Specifies the number of states considered in a surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
RECOMMENDATION:
Any states which may come close in energy to the active surface should be included in the surface hopping calculation.
FSSH_INITIALSURFACE
Specifies the initial state in a surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
None
OPTIONS:
An integer between FSSH_LOWESTSURFACE and FSSH_LOWESTSURFACE
FSSH_NSURFACES
.
RECOMMENDATION:
None
AFSSH
Adds decoherence approximation to surface hopping calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Traditional surface hopping, no decoherence.
1
Use augmented fewest-switches surface hopping (AFSSH).
RECOMMENDATION:
AFSSH will increase the cost of the calculation, but may improve accuracy for some systems. See Refs. Subotnik:2011a,Subotnik:2011b,Landry:2012 for more detail.
AIMD_SHORT_TIME_STEP
Specifies a shorter electronic time step for FSSH calculations.
TYPE:
INTEGER
DEFAULT:
TIME_STEP
OPTIONS:
Specify an electronic time step duration of
a.u.. If
electronic wave function will be integrated multiple times per nuclear time step,
using a linear interpolation of nuclear quantities such as the energy gradient and
derivative coupling. Note that
RECOMMENDATION:
Make AIMD_SHORT_TIME_STEP as large as possible while keeping the trace of the density matrix close to unity during long simulations. Note that while specifying an appropriate duration for the electronic time step is essential for maintaining accurate wave function time evolution, the electronic-only time steps employ linear interpolation to estimate important quantities. Consequently, a short electronic time step is not a substitute for a reasonable nuclear time step.
FSSH_CONTINUE
Restart a FSSH calculation from a previous run, using the file 396.0. When this is enabled, the initial conditions of the surface hopping calculation will be set, including the correct wave function amplitudes, initial surface, and position/momentum moments (if AFSSH) from the final step of some prior calculation.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
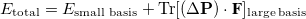
Start fresh calculation.
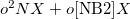
Restart from previous run.
RECOMMENDATION:
None
10.4.2 Examples
Example 10.215 Note that analytic derivative couplings must be requested via CIS_DER_COUPLE, but it is unnecessary to include a $derivative_coupling section. The same is true for SET_STATE_DERIV, which will be set to the initial active surface automatically. Finally, one must be careful to choose a small enough time step for systems that have energetic access to a region of large derivative coupling, hence the choice for AIMD_TIME_STEP_CONVERSION and TIME_STEP.
$molecule
0 1
C -1.6202943174 0.3486771706 -0.0088381608
C -0.3992060321 -0.4374935803 -0.0125353734
C -0.1051933578 -1.296810349 -1.0813404976
H -0.789110437 -1.3746939832 -1.9050808396
C 1.0690167317 -2.0450541666 -1.0723045638
H 1.2924957158 -2.701157257 -1.8896869951
C 1.956240503 -1.9403247162 0.0028420512
H 2.8596808095 -2.5170196806 0.0084200135
C 1.6662591666 -1.0840653437 1.0710071801
H 2.3481047422 -1.0057650371 1.8941404183
C 0.4955421587 -0.3357019538 1.0654970372
H 0.2532879707 0.3258430965 1.8718669605
O -1.9310452906 1.1248724768 0.9117384179
H -2.2695283633 0.2278133236 -0.8656456486
$end
$rem
jobtype aimd
exchange hf
basis 3-21g
cis_n_roots 2
symmetry off
sym_ignore true
cis_singlets false
cis_triplets true
proj_transrot true
FSSH_LOWESTSURFACE 1
FSSH_NSURFACES 2 # hop between T1 and T2
FSSH_INITIALSURFACE 1 # start on T1
AFSSH 0 # no decoherence
CIS_DER_COUPLE TRUE
aimd_steps 500
time_step 14
AIMD_SHORT_TIME_STEP 2
aimd_time_step_conversion 1 # Do not alter time_step duration
aimd_print 1
aimd_init_veloc thermal
aimd_temp 300 # K
aimd_thermostat 4 # Langevin
aimd_integration vverlet
fock_extrap_order 6
fock_extrap_points 12
$end