9.10 State Following
In some circumstances, (e.g. optimizing excited state geometries) it can be important to find and follow electronically excited states of a particular character as the geometry changes. The state-following algorithm in Q-Chem is based on the overlap of the attachment/detachment densities (Section 6.12.1) at sucessive steps. Using the densities avoids any issues that may be introduced by the orbitals changing sign.
Two parameters are used to influence the choice of the electronic surface. One (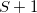 ) controls the energy window for states included in the search, and the other (
) controls the energy window for states included in the search, and the other (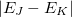 ) controls how well the states must overlap in order to be considered of the same character. These can be set by the user or generated automatically based on the magnitude of the nuclear displacement.
) controls how well the states must overlap in order to be considered of the same character. These can be set by the user or generated automatically based on the magnitude of the nuclear displacement.
The energy window is defined relative to the estimated energy (i.e. 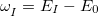 ), for the current step based on the energy, gradient and nuclear displacement of previous steps. This estimated energy is specific to the type of calculation (e.g. geometry optimization). The similarity metric for the overlap is defined as
), for the current step based on the energy, gradient and nuclear displacement of previous steps. This estimated energy is specific to the type of calculation (e.g. geometry optimization). The similarity metric for the overlap is defined as
 |
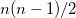 |
(9.13) | ||
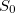 |
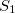 |
|||
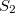 |
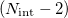 |
where 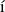 is the spectral norm of the matrix and is defined as
is the spectral norm of the matrix and is defined as 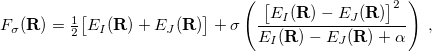 . The selected state always satisfies one of the following
. The selected state always satisfies one of the following
It is the only state in the window defined by
. It is the state with the highest overlap, provided at least one state has
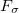 .
. It is the nearest state energetically if all states in the window have
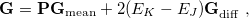 , or there are no states in the energy window.
, or there are no states in the energy window.
State following can currently be used with CIS and TDDFT excited states and is initiated with the $rem variable STATE_FOLLOW. It can be invoked for geometry optimizations, with Ab initio molecular dynamics,[490] and with the growing string method. The desired state is specified using SET_STATE_DERIV (or for FSM/GSM: SET_STATE_REACTANT and SET_STATE_PRODUCT).
The results for geometry optimizations can be affected by the step size (GEOM_OPT_DMAX) and using a step size smaller than the default value can give better results. Also, it is often challenging to converge the strings in FSM/GSM calculations.
STATE_FOLLOW
Turns on state following.
TYPE:
LOGICAL
DEFAULT:
FALSE
OPTIONS:
FALSE
Do not use state-following.
TRUE
Use state-following.
RECOMMENDATION:
None.
FOLLOW_ENERGY
Adjusts the energy window for near states
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use dynamic thresholds, based on energy difference between steps.
Search over selected state
.
RECOMMENDATION:
Use a wider energy window to follow a state diabatically, smaller window to remain on the adiabatic state most of the time.
FOLLOW_OVERLAP
Adjusts the threshold for states of similar character.
TYPE:
INTEGER
DEFAULT:
0
OPTIONS:
0
Use dynamic thresholds, based on energy difference between steps.
Percentage overlap for previous step and current step.
RECOMMENDATION:
Use a higher value to require states have higher degree of similarity to be considered the same (more often selected based on energy).